


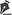
Original Article - Year 2025 - Volume 40Issue 1
Median Craniofacial Clefts: Case Series from the Center for the Treatment and Rehabilitation of Cleft Lip and Palate and Craniofacial Deformitiesx
Fissuras medianas craniofaciais: Série de casos do Centro de Tratamento e Reabilitação de Fissuras Labiopalatais e Deformidades Craniofaciais
André Luiz Monteiro dos Santos Marins1 ; Mateus Teixeira Alfenas1; Aurélio Rocha Batista de Oliveira1; Thaís Moreira1; Bruno Meilman Ferreira1; Mariana Sisto Alessi1; Júlia Cardoso Laluce1; Hugo Leonardo de Resende Rodrigues1
; Mateus Teixeira Alfenas1; Aurélio Rocha Batista de Oliveira1; Thaís Moreira1; Bruno Meilman Ferreira1; Mariana Sisto Alessi1; Júlia Cardoso Laluce1; Hugo Leonardo de Resende Rodrigues1
ABSTRACT
Introduction Walter Dick reported the first case of oblique facial cleft in 1837 and, since then, numerous cases have been described. This is a rare congenital malformation with diverse clinical manifestations, often associated with other congenital anomalies. Surgical treatment of these clefts is challenging due to the complexity and variation in presentations.
Materials and Methods The present retrospective descriptive study was conductedat the Center for the Treatment and Rehabilitation of Cleft Lip and Palate and Craniofacial Deformities (Centro de Tratamento e Reabilitação de Fissuras Labiopalatais e Deformidades Craniofaciais, CENTRARE, in Portuguese), Hospital da Baleia, between January 2006 and March 2023. We analyzed data from the medical records of 12 (33.3% of male and 66.6% of female) patients with median cleft. Variables included sex, pregnancy risk factors, family history, associated anomalies, age at first surgical procedure, cleft presentation, and surgical technique.
Results Pregnancy risk factors were identified in 58.33% of cases, and 91.66% had associated anomalies, the most common being holoprosencephaly. The age at the first surgical procedure ranged from 6 months to 1 year. Also, two patients died due to severe malformations.
Conclusion Median facial clefts present significant phenotypic variations, requiring individualized surgical approaches. Treatment planning should consider the severity of anomalies and life expectancy, prioritizing anatomical restructuring and preservation of essential functions.
Keywords: cleft lip; cleft palate; congenital hypothyroidism; face; surgery; plastic
RESUMO
Introdução Walter Dick relatou em1837, o primeiro caso de fissura facial oblíqua e, a partir desta época, inúmeros casos foram descritos. São malformações congênitas raras que apresentam diversas manifestações clínicas, frequentemente associadas a outras anomalias congênitas. O tratamento cirúrgico dessas fissuras é um desafio devido à complexidade e variação das apresentações.
Materiais e Métodos O presente estudo é de caráter retrospectivo descritivo realizado no Centro de Tratamento e Reabilitação de Fissuras Labiopalatais e Deformidades Craniofaciais (CENTRARE), Hospital da Baleia, entre janeiro de 2006 e março de 2023. Foram analisados dados de prontuários de 12 pacientes com fissura mediana, sendo 33,3% do sexo masculino e 66,6% do sexo feminino. As variáveis incluíram sexo, fatores de risco na gestação, histórico familiar, anomalias associadas, idade no primeiro procedimento cirúrgico, apresentação da fissura e técnica cirúrgica.
Resultados Fatores de risco gestacionais foram identificados em 58,33% dos casos, e 91,66% apresentaram anomalias associadas, sendo holoprosencefalia amais comum. A idade do primeiro procedimento cirúrgico variou de 6 meses a 1 ano. Além disso, dois pacientes evoluíram a óbito devido a malformações graves.
Conclusão As fissuras medianas da face apresentam variações fenotípicas significativas, exigindo abordagens cirúrgicas individualizadas. A programação do tratamento deve considerar a gravidade das anomalias e a expectativa de vida, priorizando a reestruturação anatômica e a preservação das funções essenciais.
Palavras-chave: anormalidades congênitas; cirurgia plástica; face; fenda labial; fissura palatina
Introduction
Craniofacial clefts are rare and complex congenital malformations leading to significant aesthetic, functional, and psychosocial repercussions. Walter Dick was the first to describe an oblique facial cleft in 1837 and, since then, several cases have been reported, although in a heterogeneous manner.1-3 The lack of etiological consensus has historically contributed to inconsistent descriptions and classifications of these abnormalities.4
Although the exact incidence of rare craniofacial clefts remains unknown, estimates range from 1.4 to 4.9 per 100 thousand live births.2 Etiological factors usually belong to four main categories: maternal infections, maternal metabolic disorders, radiation exposure, and use of teratogenic drugs during the first months of pregnancy.3,5
In 1976, Tessier6 proposed a detailed anatomical classification for facial clefts based on precise anatomical parameters. His numerical system ranges from 0 to 14 and includes the number 30, allowing the identification of the cleft path in soft tissues and the craniofacial skeleton. This system is a milestone in the diagnosis and therapeutic planning of these malformations.
The spectrum of median clefts is broad, ranging from subtle notches in the vermilion of the lip to complete cleft involving bony and soft tissue structures. DeMyer classified these abnormalities into two major groups according to their association withhypoteleorbitismor hyperteleorbitism.7 However, there are also rare reports of median cleft lip with normotelorism.
There are scarce reports on median cleft surgical treatment. A thorough investigation of other associated malformations, which are frequently present, is mandatory, as they can severely compromise the child’s overall development. It is crucial to carefully consider these conditions during surgical planning due to their anatomical and functional complexity.
Objective
The current study aims to analyze the different clinical presentations, epidemiological profiles, surgical management, and outcomes of patients with median facial clefts treated at a craniofacial referral center to assist in the standardization of the diagnostic and therapeutic approach.
Materials and Methods
The present retrospective, observational, and descriptive study was conducted at the Center for Treatment and Rehabilitation of Cleft Lip and Palate and Craniofacial Deformities (Centro de Tratamento e Reabilitação de Fissuras Labiopalatais e Deformidades Craniofaciais, CENTRARE, in Portuguese), Hospital da Baleia, in Belo Horizonte, Minas Gerais, Brazil. CENTRARE is a state reference center for the care of patients with craniofacial malformations. The Ethics and Research Committee of the institution approved this study (CAAE 71687023.7.0000.5123).
The study included all patients diagnosed with a median facial cleft, according to the Tessier classification, treated from January 2006 to March 2023. We excluded patients with other types of craniofacial clefts.
We collected data by analyzing medical records. The variables assessed included demographic data, pregnancyrelated factors, family history, genetic factors, clinical characteristics, and surgical aspects. The choice of surgical approach was tailored per cleft location and extent, affected anatomical structures, clinical prognosis, tissue availability, and presence of associated malformations.
The data was organized in an electronic spreadsheet and analyzed it using descriptive statistics. We calculated absolute and relative frequencies for categorical variables, and measures of central tendency and dispersion for continuous variables, such as age.
Results
This case series had 12 patients, including 33.3% male (n ¼ 4) and 66.6% female subjects (n ¼ 8).
Risk factors for the development of congenital malformations during pregnancy were present in 58.33% of cases (n ¼ 7), including 25% (n ¼ 3) with maternal metabolic disorders (gestational diabetes), 25% (n ¼ 3) with medication use, and 8.33% (n ¼ 1) with parental consanguinity. A family history of cleft lip and palate was observed in 33.3% (n ¼ 4). ►Table 1 shows the demographic characteristics and risk factors of the cohort.
| Patient | Sex | Gestational risk factor | Family history of cleft |
|---|---|---|---|
| GVOM (†6 years old) | Female | No | No |
| GHFG | Male | No | No |
| JMRN | Male | Yes (gestational diabetes) | No |
| VGSS (†2 years old) | Female | Yes (parental consanguinity) | No |
| THSB | Male | Yes (use of anti-inflammatory and antibiotics) | Yes |
| MEVS | Female | Yes (use of benzodiazepine and antidepressant) | Yes |
| MACC | Female | No | Yes |
| MCCBR | Female | Yes (gestational diabetes) | No |
| LVLD | Female | No | Yes |
| EMCC | Female | Yes (gestational diabetes) | No |
| ANAS | Female | No | No |
| MPCD | Male | Yes (use of antidepressants) | No |
Note: †Death.

Among all patients included inthestudy, 91.66% (n ¼ 11) had associated abnormalities. The cleft presentation and associated abnormalities were diverse (►Table 2). It is worth noting that the most commonly found anomaly was complex brain malformation resulting from incomplete prosencephalon cleavage (holoprosencephaly), with a total of 41.66% cases (n ¼ 5).
| Patient | Associated abnormalities | Age at first surgery | Cleft presentation | Surgery performed |
|---|---|---|---|---|
| GVOM (†6 years old) |
Holoprosencephaly, microcephaly, hypotelorism, West syndrome, heart disease, pyelocalyceal ectasia, absence of premaxilla. | 6 months | Median cleft lip and palate | Cheiloplasty |
| GHFG | Absence of premaxilla, appendix in palate, ectrodactyly, ectodermal dysplasia, and cleft lip and/or palate (EEC) syndrome, presence of six fingers on each hand, holoprosencephaly. | 11 months | Median cleft lip and palate | Cheiloplasty |
| JMRN | Hypotelorism, holoprosencephaly, and microcephaly. | None | Median cleft lip and palate | None |
| VGSS (†2 years old) |
Holoprosencephaly, microcephaly, and agenesis of the nasal bones. | None | Median cleft lip | None |
| THSB | Holoprosencephaly and West syndrome. | 6 months | Median cleft lip and palate | Cheiloplasty |
| MEVS | Absence of nasal septum and premaxilla. | None | Median cleft lip and palate | None |
| MACC | Pharyngeal appendix, agenesis of the corpus callosum, and malformation of the sella turcica and pituitary gland. | 10 months | Median cleft lip and palate | Cheiloplasty |
| MCCBR | Bilateral stenosis of the nasal cavities and double labial frenum. | 7 months | Median cleft lip with midline alveolar defect þ distal division of the nasal septum | Cheiloplasty |
| LVLD | Telecanthus, hypertelorism, tongue appendage, and bifid nose with 4 nostril holes. | 12 months | Bone deformity in the midline frontonasal region and premaxilla | Correction of the lip brace þ nasal reconstruction with full use of existing tissues |
| EMCC | Telecanthus, hypotelorism, and microcephaly. | None | Median cleft lip and palate with absence of premaxilla and vomer bone. |
None |
| ANAS | None | 11 months | Median cleft lip | Cheiloplasty |
| MPCD | Hypertelorism, telecanthus, absence of nasal bone, deviated nasal septum, heart disease, and diastema. | 1 year | Midline frontonasal deformity with bifid nose. | Nasal reconstruction |
Note: † Death.

The age at the first surgical approach ranged from 6 months to 1 year. A total of 3 patients (25%) did not undergo the procedure due to the presence of associated severe malformations, and 1 patient had not yet reached the minimum age needed during the study period. Furthermore, 2 patients (16.6%) died; one underwent cheiloplasty at 6 months died at age 6 and the other did not undergo any surgical procedure.
Discussion
Fogh-Andersen8 reported an incidence of one rare for 300 common clefts. The most affected subjects are males and Caucasians. Furthermore, 75% of patients with rare facial clefts present other abnormalities.7 In our study, these statistics were divergent, as most patients were females and 91.66% of cases had other associated malformations.
The development mechanism of Tessier’s median clefts is not fully understood, but they appear around the third week of gestation.9
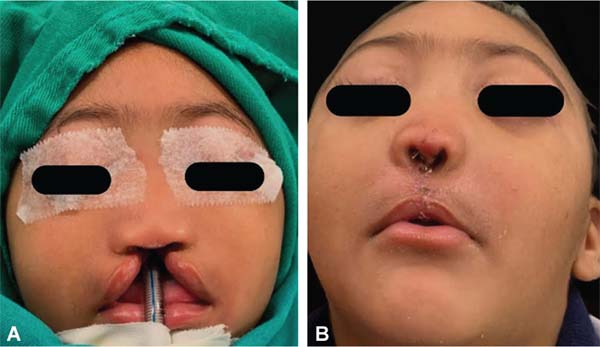
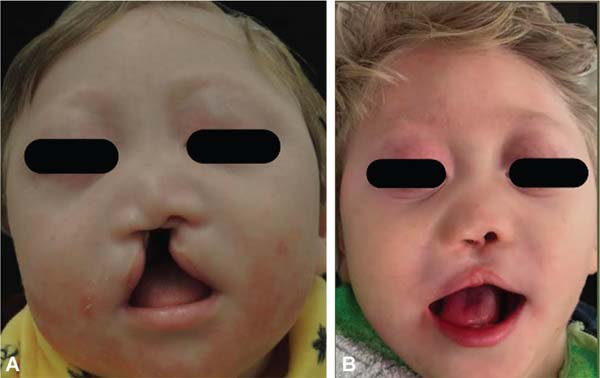
During embryogenesis, there is a failure to fuse the two medial nasal processes, potentially leading to a minimal notch in the upper lip and vermilion and a slight bifidity in the nasal tip (►Figs. 1-2) up to agenesia (►Fig. 3) or complete division and duplication of midfacial structures.10 The median cleft (Tessier’s 0) can be true11 or false.12 A true median cleft occurs between the median globular processes; the lip appears as an enlarged band, with double labial frenum (►Fig. 4) and diastema between the incisors (►Fig. 5).6 The observation of a wide columella, laterally displaced alar cartilages, and a bifid nasal tip is common. The nasal bones undergo lateral displacement. Shortening of the central height of the face and orbital hypertelorismare present,6 as can be seen in ►Fig. 6.
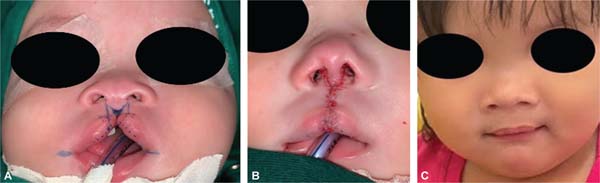
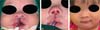
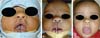
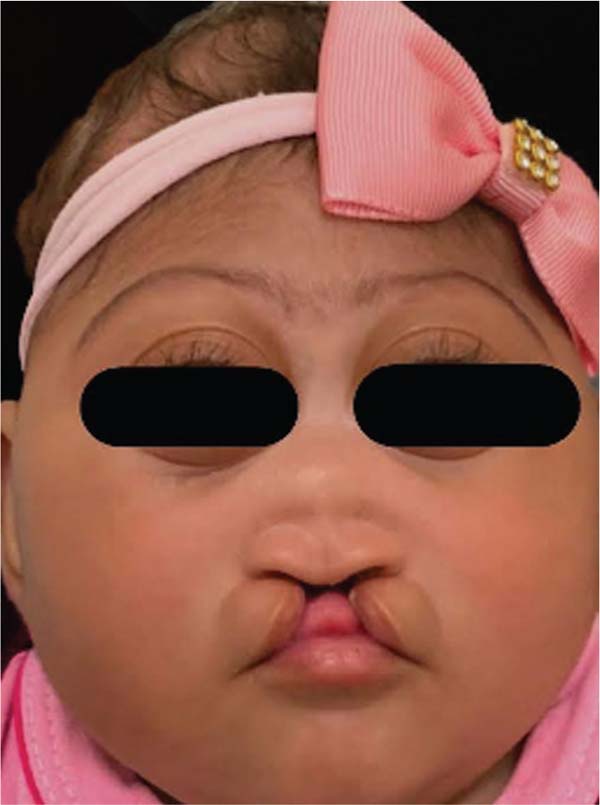

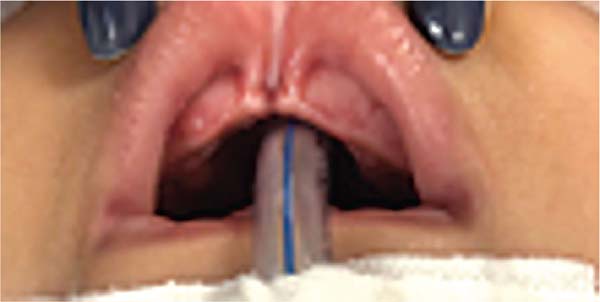
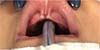
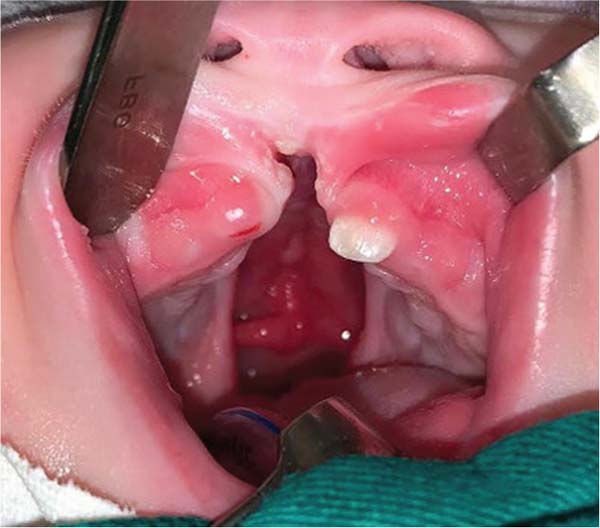
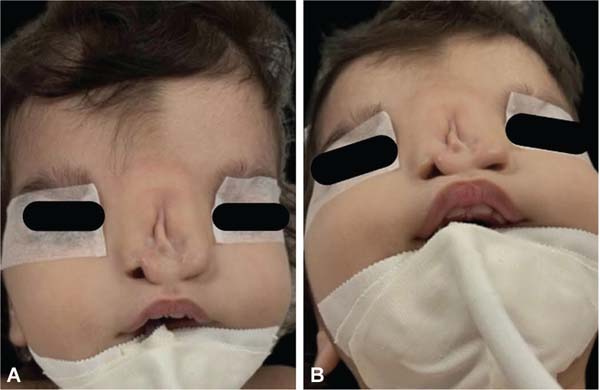
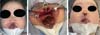
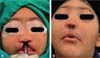
The false median cleft may present with an almost total absence of the philtrum and premaxilla, extending to the floor of the nose. Moreover, the columella does not form or is rudimentary with varied presentations (►Figs. 7-9). A cleft palate may be present (►Fig. 5), in addition to ocular abnormalities, brain anomalies, and absence of cranial integument. Bone deficiency results in hypotelorism or cyclopia.6
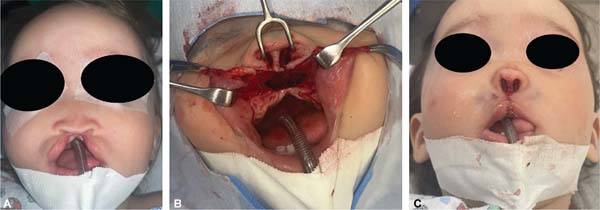

Tessier’s cleft 14 features cranial abnormalities with marked hyperor hypoteleorbitism, forming a midline craniofacial dysraphism. It can cause agenesis of a cranial segment or excessive tissue. Hypertelorism laterally displaces the orbits. Cranial and nervous system abnormalities are extreme and can often limit the life of patients.6,13 In our study, 16.6% (n ¼ 2) patients died with severe cranial anomalies.
These several facial phenotypic variations also affect the nasal anatomy, including duplicated or absent nasal septum, short and wide columella, flat dorsum, agenesis or separation of nasal bones, and nasal tips with no projection or definition. This diverse presentation requires individualized surgical approaches. Patients’ age at first surgery also depends on the severity and prognosis of each case.10,14 In clefts with significant nasal agenesis, surgical intervention of the lips and nose is recommended after 12-months-old, when the baby begins to adopt an oral breathing pattern. In cases with associated holoprosencephaly, surgical intervention after 1-year-old is also recommended due to the life expectancy of these patients.
Proper structural development improves the anatomical shape of facial components, facilitating repairs and allowing greater accuracy in approximations. In the first stage, corrections are limited to the soft tissues. The correction of deformed bone structures occurs later, according to each patient and their prognosis,14,15 waiting for facial skeleton growth.
In our study, 33.3% of patients (n ¼ 4) had a family history of cleft lip and palate, which reinforces the potential genetic influence on the etiology of the condition. This finding is consistent with data from other reference centers for clefts in the national literature, as evidenced in a study by the Hospital de Reabilitação de Anomalias Craniofaciais from Universidade de São Paulo indicating that 32.3% of patients reported the presence of at least one other subject with cleft in the family.16 It is noteworthy that the most severe forms of cleft appear to have a greater hereditary influence compared with less complex types, although this association still requires further investigation for confirmation.
Our results showed a direct correlation was observed between the severityof the craniofacial cleft and the qualityof the postoperative aesthetic outcome, which was inversely proportional to the severity of the cleft. These findings are consistent with evidence from other Brazilian centers specialized in cleft treatment,17 like the Hospital de Pediatria Professor Heriberto Bezerra from Universidade Federal do Rio Grande do Norte, and international institutions, such as the Burns & Plastic Surgery Center, Hayatabad Medical Complex, in Peshawar, and Saidu Hospital, Saidu Medical College, in Swat, both in Pakistan.18
Conclusion
As median clefts are rare and have multiple presentations, their treatment is a challenge, employing a wide range of resources due to the peculiar characteristics of each case. Thei planning is individualized, always considering the severity and prognosis of each case and surgical risks and benefits. Surgical intervention should be guided by facial anatomy restructuring by the reconstruction of nasal floor components, lip contour, and nasal projection, sparing the functions of feeding and breathing. Multidisciplinary care with psychologists, nutritionists, speech therapists, and professionals from dentistry, neurosurgery, otorhinolaryngology, and plastic surgery is essential for therapeutic success and treatment continuity.
References
1. Dick W. A case of hyperencephalous monstrouosity. Lond Med Gaz. 1837;19:897
2. Kawamoto H. Rare craniofacial clefts. In: McCarthy JG, ed. Plastic Surgery Volume 4. Philadelphia, PA: WB Saunders; 1990:2922-2973
3. Ozaki W, Kawamoto HK Jr. Craniofacial clefting. In: Lin KY, Ogle RC, JA Jane, ed. Craniofacial Surgery: Science & Surgical Technique. Philadelphia, PA: WB Saunders; 2002
4. Washington GN, Cepeda A Jr, Moffitt J, Groff CK, Greives MR, Nguyen PD. Is the Message Clear? Evaluation of Readability Levels for Cleft Lip, Cleft Palate, and Craniofacial Websites. Cleft Palate Craniofac J 2023;60(12):1619-1624. Doi: 10.1177/10556656221112672
5. Carreirão S, Lessa S, Zanini SA. Tratamento das fissuras labiopalatais. 2nd ed. Rio de Janeiro: Revinter; 1996
6. Tessier P. Anatomical classification facial, cranio-facial and laterofacial clefts. J Maxillofac Surg 1976;4(02):69-92. Doi: 10.1016/ s0301-0503(76)80013-6
7. DeMyer W. The median cleft face syndrome. Differential diagnosis of cranium bifidum occultum, hypertelorism, and median cleft nose, lip, and palate. Neurology 1967;17(10):961-971. Doi: 10.1212/wnl.17.10.961
8. Fogh-Andersen P. Rare clefts of the face. Acta Chir Scand 1965; 129:275-281
9. Mazzola RF, Mazzola IC. Facial clefts and facial dysplasia: revisiting the classification. J Craniofac Surg 2014;25(01):26-34. Doi: 10.1097/SCS.0b013e3182a2ea94
10. Nam SM, Kim YB. The Tessier number 14 facial cleft: a 20 years follow-up. J Craniomaxillofac Surg 2014;42(07):1397-1401. Doi: 10.1016/j.jcms.2014.03.032
11. Galanti S. Rare congenital malformation of the nose. (2 cases of bifid nose). Ann Laringol Otol Rinol Faringol 1961;60: 583-601
12. Braithwaite F, Watson J. A report on three unusual cleft lips. Br J Plast Surg 1949;2(01):38-49. Doi: 10.1016/s0007-1226(49) 80007-5
13. Spataro EA, Most SP. Tongue-in-Groove Technique for Rhinoplasty: Technical Refinements and Considerations. Facial Plast Surg 2018;34(05):529-538. Doi: 10.1055/s-0038-1670647
14. Lage RR, Araújo GKM, Heitor BS, Oliveira LGL. Tessier 0-14 Cleft: Surgical Management Report of One Atypical Case. Rev Bras Cir Plást 2008;23:58-60
15. Palukuri L, Sharada RJ, Naidu DV. Tessier 30 Facial Cleft: A Rare Craniofacial Anomaly. Int J Clin Pediatr Dent 2023;16(01): 177-179. Doi: 10.5005/jp-journals-10005-2506
16. Souza-Freitas JAd, Dalben GdS, Freitas PZ, Santamaria M Jr. Tendência familial das fissuras lábio-palatais. Rev Dent Press Ortodon Ortop Facial 2004;9(05):74-78. Doi: 10.1590/S1415-54192004000500009
17. Spencer LSdB, Buzzo CL. Primary treatment of lip and nasal deformity in unilateral cleft lip or cleft lip and palate [Tratamento primário da deformidade labial e nasal em fissura labial unilateral ou fissura labiopalatina]. Rev Bras Cir Plást 2017;32:37-45. Doi: 10.5935/2177-1235.2017RBCP0006
18. Khan M, HidayatullahHayat W, Khattak DA, Khan A, Hayat N, etal. Rare craniofacial clefts: Surgical management protocols. J Plast Reconstr Aesthet Surg 2024;97:41-49. Doi: 10.1016/j.bjps.2024.07.043
1. Fundação Benjamin Guimarães, Hospital da Baleia, Belo Horizonte, MG, Brazil
Address for correspondence André Luiz Monteiro dos Santos Marins, Rua Carolina Figueiredo 62, apto. 403, Serra, Belo Horizonte, MG, CEP: 3022-0130, Brazil (e-mail: dr.andremarins@gmail.com).
Artigo submetido: 17/10/2024.
Artigo aceito: 20/05/2025.
Conflict of Interests
The authors have no conflict of interests to declare.
 Read in Portuguese
Read in Portuguese
 Read in English
Read in English
 PDF PT
PDF PT
 Print
Print
 Send this article by email
Send this article by email
 How to Cite
How to Cite
 Mendeley
Mendeley
 Pocket
Pocket