


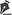
Case Report - Year 2014 - Volume 29 -
Fibromatose digital infantil: relato de caso
Infantile digital fibromatosis: case report
RESUMO
INTRODUÇÃO: A fibromatose digital infantil é uma proliferação nodular, assintomática, rara e benigna do tecido fibroso, que ocorre quase exclusivamente na região dorsal e lateral dos dedos das mãos e pés. O artigo relata um caso de fibromatose digital infantil, também conhecida como tumor de Reye.
RELATO DE CASO: Trata-se de um caso diagnosticado por meio de exames clínico, de imagem e histopatológico. O paciente apresentou-se ao Setor de Ortopedia do Hospital, queixando-se de uma lesão nodular, eritematosa, indolor, no segundo pododáctilo do pé esquerdo, existente havia quatro anos. Durante o exame físico, notava-se uma deformidade no II pododáctilo, causada por uma lesão nodular, eritematosa, indolor, de aproximadamente 1,5cm, que não acarretava alterações funcionais. O exame de ultrassom revelou a presença de uma imagem nodular sólida, hipoecogênica, envolvendo o tendão do extensor do II pododáctilo na falange média. O diagnóstico inicial era de fibroma ou sinovioma. Pelas características clínicas da lesão, por seu tempo de evolução e pelos achados de imagem, a equipe optou por uma biópsia. No entanto, devido ao pequeno tamanho da lesão, sendo a biópsia aberta, realizou-se a exérese cirúrgica. O exame histopatológico confirmou o diagnóstico de fibromatose digital infantil.
CONCLUSÃO: Esse tumor constitui uma entidade clínica rara, que deve ser diferenciada de outras lesões encontradas nos dedos das mãos e dos pés. O diagnóstico correto raramente é feito antes da operação, devido, principalmente, à falha em reconhecer essa entidade. Por essa razão, é essencial considerar essa lesão em diagnósticos diferenciais.
Palavras-chave: Pé; Fibromatose digital infantil; Tumor de Reye; Relato de caso.
ABSTRACT
INTRODUCTION: Infantile digital fibromatosis, also known as Reye tumor, is a rare, asymptomatic, benign nodular proliferation of fibrous tissue, which occurs almost exclusively in the dorsolateral region of the fingers and toes. This article reports a case of infantile digital fibromatosis.
CASE REPORT: This case was diagnosed by clinical, imaging, and histopathological examination. The patient presented at the Orthopedic Department of our hospital, with a 4-year history of a painless, erythematous nodular lesion on the second toe of the left foot. On physical examination, a deformity of the second toe caused by a nodular, erythematous, painless lesion of approximately 1.5 cm diameter was noted; the lesion did not result in functional changes. Ultrasound examination revealed a solid, hypoechoic nodule involving the extensor tendon in the middle phalanx of the second toe. The initial diagnosis was fibroma or synovioma. Due to the clinical characteristics of the lesion, its evolution, and the imaging findings, the team chose to perform a biopsy. However, due to the small size of the lesion, upon open biopsy, surgical excision was performed. Histopathological examination confirmed the diagnosis of infantile digital fibromatosis.
CONCLUSION: Infantile digital fibromatosis is a rare clinical entity, which should be differentiated from other lesions found in the fingers and toes. The correct diagnosis is rarely made pre-operatively, due mainly to a failure to recognize this entity. For this reason, it is essential to onsider this lesion in the differential diagnosis of digital nodules.
Keywords: Foot; Infantile digital fibromatosis; Reye tumor; Case Report.
O conceito de fibromatose foi estabelecido por Stout, constituindo um importante avanço no estudo dos tumores de partes moles1. As fibromatoses englobam amplo espectro de proliferações benignas do tecido fibroso, com achados histológicos semelhantes e comportamento biológico intermediário entre lesões benignas e malignas. Essas neoplasias são tumores não capsulados capazes de infiltrar estruturas adjacentes, porém sem nunca haver ocorrência de metástases regionais e/ou sistêmicas. Atualmente, a classificação proposta por Enzinger & Weiss é a mais aceita e utilizada: fibromatoses superficiais (fasciais) e fibromatoses profundas (musculoaponeuróticas)2.
As fibromatoses superficiais são frequentemente pequenas e apresentam crescimento lento. Elas incluem a fibromatose palmar e a plantar, o fibroma aponeurótico juvenil e a fibromatose digital infantil. As fibromatoses profundas são grandes e podem aumentar rapidamente. Elas são mais agressivas e provavelmente crescem da fáscia profunda sobre o músculo e o tecido aponeurótico; esse grupo inclui a miofibromatose infantil, a fibromatose colli, o tumor desmoide extra-abdominal e a fibromatose agressiva infantil.
A fibromatose digital infantil é uma lesão rara e benigna, mais frequente nos dedos das mãos e pés, e que acomete principalmente as regiões dorsal e lateral3-13. Essa lesão foi primeiramente descrita por Reye, em 1965, como um tumor fibroso digital recorrente7. Segundo Werther e Seiersen (1997), foram descritos aproximadamente 100 relatos dessa patologia na literatura3. A despeito desses 100 casos, ainda não há um consenso sobre o tratamento ideal disponível na literatura1-13.
A origem dessa patologia é desconhecida, não existindo prevalência de sexo atingido ou tendência familiar. Em um terço dos casos, a lesão está presente desde o nascimento3. A fibromatose digital infantil, devido à sua raridade, é pouco conhecida nos meios ortopédicos e dermatológicos. Por isso, é essencial considerar essa condição em diagnósticos diferenciais de lesões que acometam pés e mãos de crianças para se evitar condutas equivocadas. Essa lesão tende a regredir, após dois ou três anos de evolução, em aproximadamente 12% dos pacientes e, em torno de 60% dos casos cirúrgicos, apresenta recidiva3-4, 10,12-13.
O relato feito neste trabalho descreve um caso de fibromatose digital infantil no segundo pododáctilo esquerdo em um paciente de cinco anos de idade, diagnosticado por meio de exames clínico, de imagem e histopatológico.
RELATO DO CASO
O paciente HZR, sexo masculino, leucodérmico, cinco anos de idade, apresentou-se ao Setor de Ortopedia do Hospital, em 28 de agosto de 2004, com queixa de aparecimento de lesão nodular no segundo pododáctilo esquerdo. Na anamnese não foram encontrados história de trauma prévio, comorbidades ou dados que pudessem auxiliar no diagnóstico. Em exame físico, apresentava lesão nodular, eritematosa, de aproximadamente 1,5cm de diâmetro, indolor, na região dorsolateral do segundo pododáctilo esquerdo, entre a falange média e a distal (Figura 1). A lesão não acarretava alterações funcionais do dedo.
Figura 1. Paciente apresentando lesão nodular de aproximadamente 1,5cm de diâmetro na região dorsal e lateral do segundo pododáctilo do pé esquerdo.
Solicitada a ultrassonografia, verificou-se a presença de imagem nodular sólida, hipoecogênica, envolvendo o tendão extensor do segundo pododáctilo esquerdo, ao nível da falange média, medindo 12 x 8,2 x 4,7mm de diâmetro longitudinal, transverso e anteroposterior, respectivamente. A hipótese diagnóstica era de fibroma ou sinovioma.
Pelas características clínicas da lesão, por seu tempo de evolução (aproximadamente quatro anos) e pelos achados de imagem, a equipe optou por biópsia. No entanto, devido ao pequeno tamanho da lesão, sendo a biópsia aberta, realizou-se a exérese cirúrgica, com margens de +-3mm, por meio de bloqueio anestésico local associado a sedação e fechamento primário da lesão com sutura simples.
Solicitado o histopatológico da peça, esta evidenciou ao exame macroscópico: elipse de pele medindo 1,5x0,5 x0,4cm e apresentando área elevada na superfície cutânea. Cortes a esse nível mostram tecido firme e esbranquiçado. Acompanham fragmentos de tecido também firme e esbranquiçado, medindo em conjunto 1,5x1,5x0,5cm. A microscopia revelou cortes histológicos de fragmento de pele apresentando a epiderme conservada. Na derme, observou-se proliferação de fibroblastos circundados por estroma colágeno denso. Essas células se dispõem em feixes, que se estendem da epiderme às camadas profundas da derme, formando nódulo mal delimitado. A coloração pelo Tricômero de Masson revelou inclusões citoplasmáticas pequenas e eosinofílicas próximas ao núcleo dos fibroblastos (figura 2). O patologista concluiu o laudo afirmando que os achados eram compatíveis com fibromatosedigital infantil.
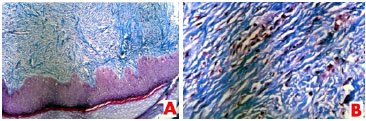
Figura 2. Fotomicrografia (A): Lâmina histológica revelando tecido epidérmico conservado com proliferação fibroblástica difusa na derme. Coloração pelo Tricômero de Masson (100x). (B): Fotomicrografia demonstrando inclusões citoplasmáticas pequenas e eosinofílicas situadas próximo aos núcleos dos fibroblastos. Coloração pelo Tricômero de Masson (400x).
Após a cirurgia, o paciente foi orientado a retornar ao setor de ortopedia para controle. No retorno, em fevereiro de 2005, aproximadamente cinco meses após a cirurgia, verificou-se a formação de uma tumoração com as mesmas características clínicas da lesão anterior. Foi solicitada ultrassonografia, no dia 15 de fevereiro de 2005. O exame revelou imagem nodular sólida, hipoecogênica, envolvendo o tendão extensor do segundo pododáctilo esquerdo, na falange média. Concluindo-se pela recidiva da lesão - fibromatose digital infantil - optou-se pela conduta expectante, orientando a família sobre a benignidade da lesão e a probabilidade de regressão espontânea.
DISCUSSÃO
A fibromatose digital infantil, também conhecida como tumor de Reye, é um tumor raro e benigno. Esse tipo de fibroma difere dos outros tipos em três aspectos: é limitado aos dedos das mãos e pés de crianças, demonstra uma tendência notável à recorrência (em mais de 60% dos casos) e em exame histopatológico apresenta corpos de inclusão citoplasmáticos3,4,6,7. Essa lesão foi primeiramente descrita por Reye, em 1965, como um tumor fibroso digital recorrente5.
A etiologia dessa doença é desconhecida. Uma infecção viral foi inicialmente considerada suspeita pela presença de corpos de inclusão citoplasmáticos encontrados em exame histopatológico, mas os achados da microscopia eletrônica e da reação em cadeia de polimerase não sustentaram essa etiologia9.
Do ponto de vista embriológico, o mesoderma dá origem aos mesoblastos e, a partir daí, aos pré-fibroblastos e fibroblastos. O pré-fibroblasto, dependendo dos fatores de influência, pode se diferenciar em uma grande variedade de células do tecido conjuntivo, que inclui fibroblastos, mioblastos, lipoblastos e, inclusive, em outros tipos de células, tais como osteoblastos, condroblastos, células sanguíneas e linfoblastos. O fibroblasto merece particular atenção por possuir um alto potencial oncogênico, podendo dar origem a variedades de tumores que vão desde a benigna fibromatose digital infantil até a mortal fibromatose agressiva, encontrando-se entre esses extremos, outros tumores como histiocitoma fibroso maligno, miofibromatose infantil, tumor desmoide, fibrosarcoma e outros9.
Os fatores que incidem no potencial oncogênico dos fibroblastos são múltiplos: genéticos, ligados ao sexo, hormonais, de crescimento e outros vários, como foi sugerido na fibromatose digital infantil pela presença de corpos de inclusão citoplasmáticos, apesar dessa possibilidade não ter sido confirmada. Presume-se também que a circulação de hormônios maternos anormais no sangue do recém-nascido produza um rápido aparecimento de tumorações logo após o nascimento, e sua posterior involução espontânea9.
Os nódulos podem ser únicos ou múltiplos, de consistência firme ou gelatinosa, eritematosos, podendo atingir mais de 2cm de diâmetro e acometendo quase exclusivamente dedos dos pés e mãos, principalmente na região dorsal e lateral, poupando, na grande maioria dos casos, o polegar e o primeiro pododáctilo3-5. Existem raros relatos de lesões extradigitais, apresentando corpos de inclusão citoplasmáticos, encontradas em braços, pés, mamas e língua3-5.
A maioria dos nódulos aparece nos primeiros meses de vida, sendo que um terço está presente desde o nascimento. Aproximadamente, 75% dos casos são notados no primeiro ano de vida1-8. Relatos de acometimentos em crianças maiores ou adultos são extremamente raros. Não há prevalência de sexo, tendência familiar ou história de trauma prévio na ocorrência da fibromatose digital infantil, que é responsável por 0,1% dos tumores fibrosos encontrados em crianças5,8,10.
O diagnóstico diferencial é feito com relativa facilidade devido à localização, idade de acometimento e pela presença de corpos de inclusão citoplasmáticos, que são únicos da fibromatose digital infantil. Os diagnósticos diferenciais consistem em fibroqueratoma digital adquirido, dermatofibroma, granuloma anular, neurilemoma, sarcoidose, angiofibroma e fibrosarcoma4,5,8,10,11. A biópsia da lesão é recomendada em todos os casos para confirmação diagnóstica4,5,11,13.
Os achados histopatológicos na fibromatose digital infantil são únicos. O entrelaçamento de fascículos de células alongadas e dos feixes de colágeno forma o nódulo na derme9, o qual pode estender-se até o tecido subcutâneo. Os núcleos ovais das células são acompanhados de corpos de inclusão citoplasmáticos que correspondem a microfilamentos de actina. A epiderme sobre o nódulo pode apresentar hiperqueratose ou acantose. As estruturas anexiais e o tecido lipídico podem estar cercados por tecido fibroso proliferado, o qual, excepcionalmente, pode estender-se até o periósteo e erodi-lo, mas sem nunca acometer o osso. A imunohistoquímica é positiva para vimentina, citoqueratina, desmina e actina muscular específica, o que aponta para uma origem miofibroblástica das células. É negativa para proteína S-100 e GFAP, sendo o CD57 positivo em alguns casos9.
Em relação ao tratamento, por ser uma patologia benigna, estudos mais recentes recomendam uma conduta expectante4,7. Em aproximadamente 12% dos casos relatados, houve involução espontânea em um tempo médio de dois a três anos3,4,6,8. A cirurgia somente é recomendada em caso de deformidade ou alteração funcional3-4,6-8. De acordo com Dabney et al., dos 92 casos reportados, 78 foram tratados cirurgicamente, com 64% de recorrência e 10% de necessidade de amputação10. A cirurgia micrográfica de Mohs pode ser utilizada em caso de comprometimento funcional, embora o índice de recorrência permaneça alto11-12. Corticoides e imiquimod tópico não demonstraram nenhum benefício. Entretanto, a infiltração de corticoide na lesão pode ser benéfica. A principal droga usada é a triancinolona, na dose de 10mg/mL intralesional, um total de 0,1-0,3mL/lesão, dependendo do diâmetro. A dose pode ser repetida dentro de quatro a seis semanas, até adequada regressão da lesão3,4. Existem ainda relatos bem sucedidos de infiltração com fluorouracil13.
As principais complicações da fibromatose digital infantil consistem em deformidades, alterações funcionais e, em alguns casos, ulceração4. Essas complicações são raras e o prognóstico é excelente, pois em 12% dos casos, ocorre regressão espontânea da lesão entre dois e três anos, além de não haver relatos de malignização ou metástases na literatura consultada. Uma característica dessa patologia é a alta taxa de recorrência (aproximadamente 60%)3-5,11-13.
CONCLUSÃO
Concluindo, a fibromatose digital infantil constitui uma entidade clínica rara, que deve ser diferenciada de outras lesões nodulares encontradas nos dedos de pés e mãos. Raramente o diagnóstico correto é feito antes da cirurgia, devido principalmente à falha em reconhecer essa entidade. Por isso, é essencial considerar essa lesão em diagnósticos diferenciais para evitar condutas inadvertidas. Ressalta-se a importância da orientação aos familiares sobre a benignidade da lesão.
REFERÊNCIAS
1. Stout AP. Juvenile fibromatoses. Cancer. 1954;7(5):953-78.
2. Enzinger, FM. & Weiss, SW.: "Fibromatosis", in Soft tissue tumors, 3rd ed., St. Louis, C.V. Mosby, 1995;201-229.
3. Werther K, Seiersen M. Recurrent infantile digital fibromatosis. Ugeskr Laeger. 1997;159:4656-7.
4. Ishii N, Matsui K, Ichiyama S, Takahashi Y, Nakajima H. A case of infantile digital fibromatosis showing spontaneous regression. Br J Dermatol. 1989;121:129-33.
5. Reye RD. Recurring digital fibrous tumors of childhood. Arch Pathol. 1965;80:228-31.
6. Kawaguchi M, Mitsuhashi Y, Hozumi Y, Kondo S. A case of infantile digital fibromatosis with spontaneous regression. J Dermatol. 1998;25:523-6.
7. De León BB, Fernández VJ. Fibromatosis con cuerpos de inclusión. An Med Assoc Med Hosp ABC. 2004;49(3):147-50.
8. Baser NT, Tuncali D, Akbuga UB, Aslan G. Infantile digital fibromatosis: late results of two different treatment approaches. Eur J Plast Surg. 2006;29:38-40.
9. Zhu WY, Xia MY, Huang YF, Leonardi C, Penneys NS. Infantile digital fibromatosis: ultraestructural human papillomavirus and herpes simplex virus DNA observation. Pediatr Dermatol. 1991;8:137-9.
10. Dabney KW, MacEwen GD, Davis NE. Recurring digital fibrous tumor of childhood: case report with long-term follow-up and review of the literature. J Pediatr Orthop. 1986;6(5):612-7.
11. Albertini JG, Welsch MJ, Conger LA, et al. Infantile digital fibroma treated with Mohs micrographic surgery. Dermatol Surg. 2002;28:959-61.
12. Campbell LB, Petrick MG. Mohs micrographic surgery for a problematic infantile digital fibroma. Dermatol Surg. 2007;33:385-7.
13. Oh CK, Son HS, Kwon YW, Jang HS, Kwon KS. Intralesional fluorouracil injection in infantile digital fibromatosis. Arch Dermatol. 2005;141:549-50.
1-Ortopedista e Traumatologista especializado em Cirurgia de Mão. Membro da Sociedade Brasileira de Cirurgia de Mão. - Médico Ortopedista e Traumatologista, especialista em cirurgia de mão do Hospital Geral Governador Israel Pinheiro - IPSEMG - de Belo Horizonte e do Hospital Unimed Betim/MG
2-Cirurgião plástico, membro especialista da Sociedade Brasileira de Cirurgia Plástica (SBCP), membro do corpo clínico do Hospital Unimed, Biocor Instituto, Hospital Vera Cruz e do Instituto de Mastologia, Odontologia e Cirurgia Plástica (IMOP), Belo Horizonte, MG, Brasil
3-Ortopedista e Traumatologista do Hospital Unimed Betim/MG. Especialista em Cirurgia Artroscópica de Joelho. - Ortopedista e Traumatologista do Hospital Unimed Betim/MG
Instituição: Hospital Unimed Betim.
Autor correspondente:
Gustavo Augusto Matos Saliba
Avenida Governador Valadares, nº 454, Centro
Betim, MG, Brasil
E-mail: salibagustavo@yahoo.com.br
Artigo submetido: 23/5/2011
Artigo aceito: 29/7/2011
 Read in Portuguese
Read in Portuguese
 Read in English
Read in English
 PDF PT
PDF PT
 Print
Print
 Send this article by email
Send this article by email
 How to Cite
How to Cite
 Mendeley
Mendeley
 Pocket
Pocket