


Case Report - Year 2015 - Volume 30 -
Síndrome de Proteus: relato de casos
Proteus Syndrome: case reports
RESUMO
INTRODUÇÃO: A síndrome de Proteus é uma doença complexa e rara, classificada nos grupo das hamartoses. Foi primeiramente descrita em dois pacientes, em 1979, por Cohen e Hayden. Existe dificuldade no diagnóstico, sendo comum a confusão com síndromes de Klippel-Trenaunay-Weber, neurofibromatose ou Stuge-Weber. Apresentamos dois casos tratados no Serviço de Cirurgia Plástica e Reparadora da Universidade Federal do Paraná.
MÉTODO: Paciente masculino (caso 1), que chegou ao serviço aos 6 anos de idade, tendo como principais apresentações lipomatoses e assimetrias. A segunda paciente (caso 2) deu entrada no serviço com 20 anos de idade e diagnóstico de síndrome de Klippel-Trenaunay-Weber, que posteriormente mostrou se tratar de síndrome de Proteus.
CONCLUSÃO: A hipótese etiológica mais aceita para a doença é genética. Acredita-se que exista mosaicismo somático e que a doença seja letal no estado não mosaico. Morte prematura é bastante frequente. Entretanto, a sequela mais comum é a ocorrência de tumores incomuns. O cuidado dos pacientes portadores da síndrome é um desafio devido às suas consequências médicas e psicossociais.
Palavras-chave: Síndrome de Proteus; Hamartoma; Lipoma.
ABSTRACT
INTRODUCTION: Proteus syndrome is a complex and rare disorder classified as a hamartomatous disease. It was first described in two patients in 1979, by Cohen and Hayden. Proteus syndrome is difficult to diagnose, and is often confused with Klippel-Trenaunay-Weber syndrome, neurofibromatosis, or Sturge-Weber syndrome. In this study we describe two patients who were treated at the Plastic and Reconstructive Surgery Service of the Federal University of Paraná.
METHOD: A 6-year-old male patient (case 1) presented to the Service with lipomatosis and asymmetry, as the primary findings. A 20-year-old (case 2) was admitted to the Service with a diagnosis of Klippel-Trenaunay-Weber syndrome, which later was shown to be Proteus syndrome.
CONCLUSION: The etiological hypothesis that is most accepted for this disease is genetic. It is believed that somatic mosaicism may occur during pathogenesis, which can be lethal in the mosaic state. Premature death is common. However, the most common sequelae are the occurrence of unusual tumors. The care of patients with this syndrome is a challenge due to medical and psychosocial consequences.
Keywords: Proteus syndrome; Hamartoma; Lipoma.
A síndrome de Proteus é uma doença complexa e rara, classificada no grupo das hamartoses. Foi primeiramente descrita em dois pacientes, em 1979, por Cohen e Hayden1. Em 1983, Wiedemann et al.2 nomearam a doença como síndrome de Proteus, devido à capacidade deste Deus grego de modificar sua forma corporal, o que remete à variabilidade na expressão clínica da síndrome. A etiologia da síndrome ainda não está totalmente esclarecida, porém, acredita-se que as causas genéticas estejam envolvidas, devido a sua característica multissistêmica3. Existe dificuldade no diagnóstico da síndrome devido a sua raridade e à similaridade com outras síndromes como Klippel-Trenaunay-Weber, neurofibromatose, lipomatose encefalocraniocutânea (ECCL), Maffuci, Sturge-Weber, e Bannayan-Zonana4-7.
A apresentação da síndrome de Proteus é variada, consistindo em uma variedade de tumores de crescimento desproporcional e assimétrico em diversas partes do corpo, particularmente tumores ósseos, nevus cerebriforme de tecido conjuntivo, nevus epidermoide, malformações vasculares, e depósitos irregulares de tecido adiposo. Vários outros achados são possíveis, entre eles esplenomegalia, hipertrofia digital, escoliose, hipertrofia de lábio e dedos, cardiopatias, fibrose cística e retardo mental2,3,8,9. Os achados clínicos podem aparecer ao nascimento, porém, são mais proeminentes na evolução pós-natal destes pacientes9. Poucos trabalhos abordam as modalidades terapêuticas que podem trazer benefícios a estes pacientes. Neste trabalho, apresentamos três pacientes com diagnóstico de Síndrome de Proteus, discutimos o diagnóstico e o tratamento realizado.
RELATO DE CASOS
Caso 1
Paciente masculino, 6 anos de idade, foi encaminhado ao serviço de Cirurgia Plástica do Hospital de Clínicas devido à linfangiomas disseminados pelo dorso e membros inferiores com início aos 5 meses de idade e crescimento progressivo de volume. Nasceu de pais saudáveis, não consanguíneos, ambos com 31 anos na época da concepção, e mãe gesta III. Sua irmã mais velha faleceu duas horas após parto e seu irmão mais novo faleceu aos 3 dias de idade. Não havia outros casos semelhantes na família. Foi realizado parto cesariana na 32ª semana de gestação, não havendo relato da causa da interrupção precoce. Ao nascer, pesou 2730 gramas, ficando internado durante sete dias devido à icterícia pós-natal. Aos 4 meses de idade, submeteu-se à postectomia e correção de hérnia inguinal. Teve desenvolvimento psicomotor retardado, sentando sem apoio aos 8 meses e andando sem auxílio somente aos 2 anos.
Aos 6 anos de idade, apresentava 36,5 kg; 128,5 cm de estatura, perímetro cefálico de 55 cm, orelha com 6,5 cm, distância interpupilar com 5,5 cm, distância intermamilar com 15,5 cm, perna direita com 25 cm e perna esquerda com 28,5 cm. Observaram-se massas tumorais dorsais de consistência amolecida de grande volume; assimetria de demídeos corporais sendo maior o lado esquerdo; pé plano e hálux varo; encurtamento de 1 cm do membro inferior esquerdo e a circunferência do mesmo 1 cm maior. O exame eletrocardiográfico demonstrou arritmia sinusal. Não foram encontradas anomalias cranianas, nem sinais de retardo mental. A avaliação genética constatou cariótipo 46 XY. Teve, então, o diagnóstico de Síndrome de Proteus. (Figuras 1A e 1B).
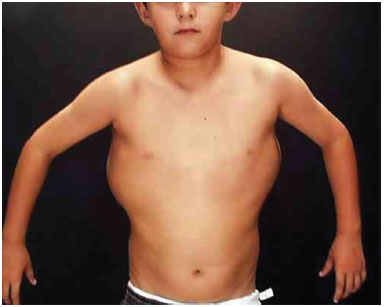
Figura 1A. Paciente apresenta linfangiomas disseminados pelo dorso.
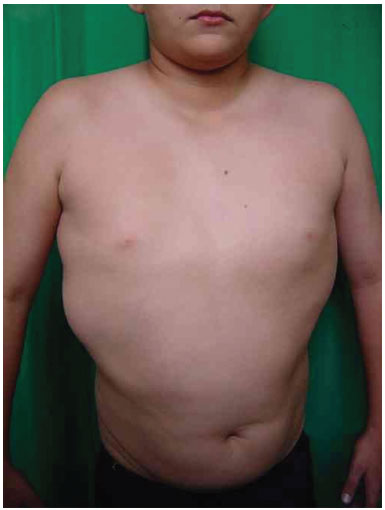
Figura 1B. Paciente apresenta linfangiomas disseminados pelo dorso.
Na ressonância magnética, identificaram-se massas tumorais acima e abaixo do músculo grande dorsal, com vasos de calibre ao redor de 0,5 cm, que nutriam a massa. Optou-se pela realização da lipoaspiração ultrassônica, por haver um relato de literatura e o menor potencial de sangramento cirúrgico. Realizou-se em abril de 2001, com retirada de 600 ml de cada lado e ressecção parcial do tecido tumoral, com boa evolução pós-operatória. Porém, permaneceram massas de grande volume em dorso, e o resultado foi considerado inadequado. Em 2003, realizou-se a segunda intervenção com ressecção de massa tumoral de 1434 gramas que englobava a musculatura do dorso à direita, sem complicações. O resultado anatomopatológico concluiu tratar-se de lipomatose (Figuras 1C e1D). Em junho de 2003, o paciente foi submetido à ressecção da massa em região dorsal à esquerda, pesando 1350 gramas. Teve também o mesmo resultado anatomopatológico. Em janeiro de 2006, o paciente foi submetido à nova ressecção das tumorações de gordura entre planos musculares, abaixo do músculo grande dorsal. O conjunto das peças pesava 175 gramas, sem alterações no exame patológico. Houve complicações pós-operatórias, com aparecimento de sinais flogísticos locais, porém, a evolução foi favorável com uso de cefalexina e analgésicos.
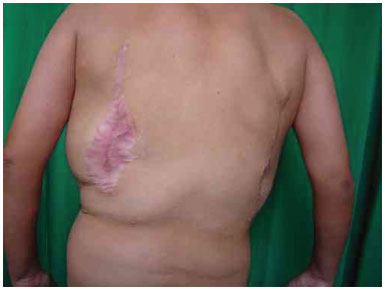
Figura 1C. Imagem do Pós operatório da ressecção da lesão.
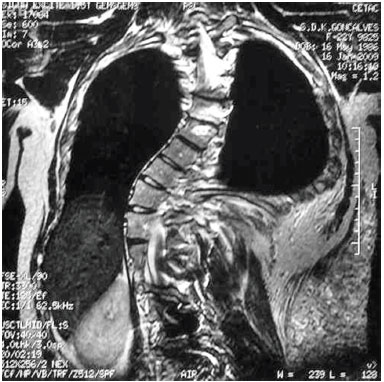
Figura 1D. Ressonância magnética demonstrando a presença de massas tumorais acima e abaixo do músculo grande dorsal.
Caso 2
Paciente feminina, encaminhada ao serviço de Cirurgia Plástica do Hospital de Clínicas de Curitiba aos 20 anos de idade pela presença de lipomas em dorso, de início aos 6 meses de idade. Pais saudáveis, não consanguíneos, a mãe com 21 anos e o pai 22 anos. Gestação sem intercorrências. Foi realizado parto cesáreo a termo. Ao nascer pesou 3100g, mediu 45 cm de comprimento e 34 cm de perímetro cefálico. Apresentou icterícia neonatal que se resolveu com fototerapia. Apresentava dois irmãos normais. Não há relato de doenças ou outros casos semelhantes na família. Ao exame, observaram-se três massas de consistência amolecida em região dorsal esquerda, móveis, indolores, não aderentes, variando de 8 a 10 cm de diâmetro. Observou-se, também, hipertrofia de membro inferior direito, com diferença de 2 cm na circunferência da coxa direita para a esquerda, e da perna direita para a esquerda. Apresentava malformação vascular tipo capilar em face lateral esquerda de tronco.
Foi primeiramente tratada pelo serviço de ortopedia por queixa de assimetria em membro inferior direito e cifoescoliose toraco-lombar tratada com artrodese e haste de Harrington. No serviço de cirurgia pediátrica foi avaliada em relação aos lipomas. O diagnóstico inicial foi de síndrome de Klippel-Trenaunay-Weber e os primeiros controles radiográficos mostraram ausência de alterações ósseas.
Nesta síndrome, a presença de mancha vinho do Porto, varizes e hipertrofia óssea a tríade está presente em 98-100% dos casos, sendo que a mancha vinho do porto e alterações vasculares geralmente estão presentes no membro hipertrofiado. A síndrome Klippel-Trenaunay-Weber é uma malformação congênita que pode decorrer de uma mutação para dominante autossômica, quando ocorrida na embriogênese, ou para autossômica dominante com penetrância incompleta. Isso explica a ocorrência familiar. Esta não apresenta um padrão de periodicidade geracional regular, podendo a síndrome ser transmitida por muitas gerações por indivíduos fenotipicamente normais10.
A avaliação genética foi baseada na anamnese. Mostrou como dados clínicos a lipomatose dorsal, malformação capilar, escoliose e assimetria de pés (sendo o lado direito maior). Por não existirem exames laboratoriais, o diagnóstico de síndrome de Proteus foi baseado em dados clínicos e familiares. Exame eletrocardiográfico demonstrou uma arritmia sinusal.
Foram realizados três procedimentos cirúrgicos para retiradas de massas tumorais dorsais. O primeiro procedimento, aos 11 anos de idade, foi lipoaspiração em dorso para a retirada dos lipomas. Pela avaliação do serviço de cirurgia plástica, foi constatada hipertrofia generalizada de tecido gorduroso. Doze anos após a primeira cirurgia, a paciente apresentou recidiva das lesões lipomatosas. Foi realizada, então, tomografia computadorizada que revelou diversas imagens císticas em hemitórax esquerdo, tubulares e apresentando fluxo vascular em seu interior, compatível com malformação venosa. Realizou-se, então, o segundo procedimento de retirada de massa de região posterior, com ressecção de peça de aspecto lobulado, de 200g, de 17 cm em seu maior comprimento, compatível com lipoma gigante ao exame anatomopatológico. A paciente foi submetida, por último, a procedimento de lipoaspiração em região de dorso. Avaliada ambulatorialmente em 2006, apresentando estabilização das massas e da malformação vascular (Figura 2 A e 2 B).
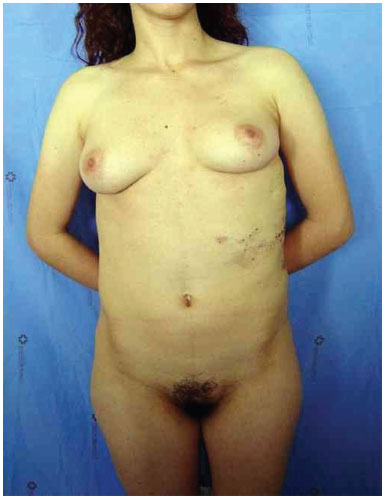
Figura 2A. Paciente com quadro de lipoamtose em tórax.
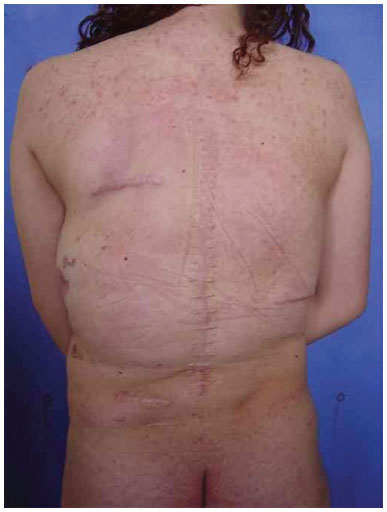
Figura 2B. Imagem do pós operatório.
Caso 3
Paciente masculino, 1 ano e 7 meses de idade, foi encaminhado ao serviço de Cirurgia Plástica do Hospital de Clínicas devido à linfangioma volumoso em região de flanco e hipocôndrio direito, estendendo-se até região dorsal. Nasceu de pais saudáveis, não consanguíneos, mãe gesta I. Não havia outros casos semelhantes na família (Figuras 3 A, B, C e D).
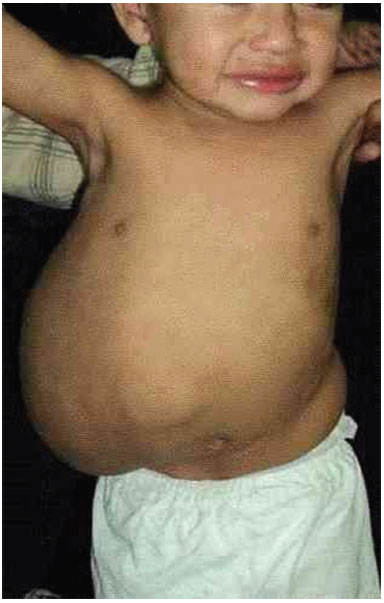
Figura 3A. Paciente com quadro de linfangioma volumoso em região de flanco e hipocôndrio direito extendendo-se até regiâo dorsal.
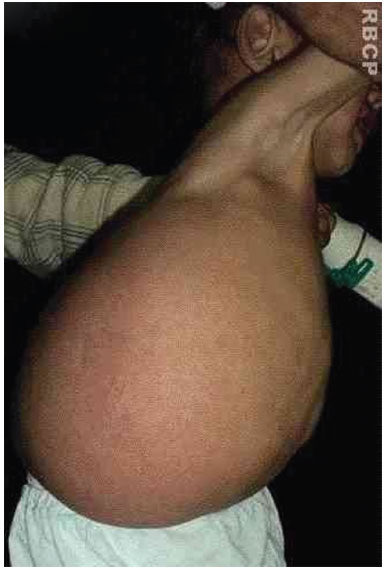
Figura 3 B. À linfangioma volumoso em região de flanco e hipocôndrio direito extendendo-se até regiâo dorsal.
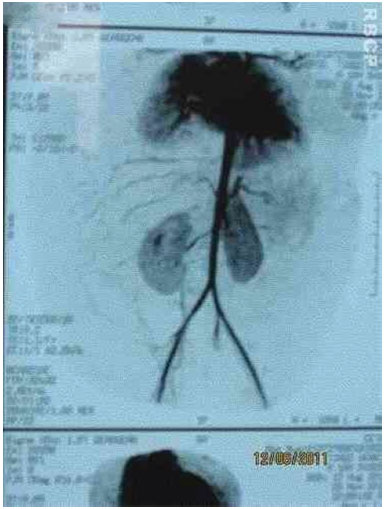
Figura 3C. Imagem da Arteriografia da lesão.
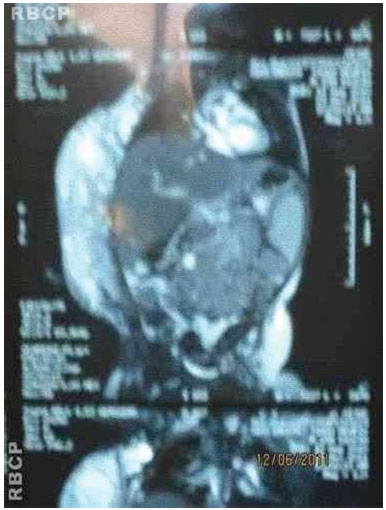
Figura 3D. Imagem da RNM da lesão tumoral.
Foi internado de emergência com choque séptico, sem apresentar um foco definido, realizado tratamento clínico com antibiótico de amplo espectro. Pressupondo-se que o foco seria o linfangioma, aguardou-se melhora do quadro clínico para a programação cirúrgica.
Foi realizada ressecção completa do tumor. Dissecção até o músculo reto abdominal, sem penetrar na cavidade abdominal, com limite superior no quinto arco costal, limite posterior à escápula e limite inferior à região inguinal. Posteriormente, foi retirado enxerto de pele total da própria massa ressecada e enxertada no mesmo ato operatório (Figura 3 E, F, G e I).
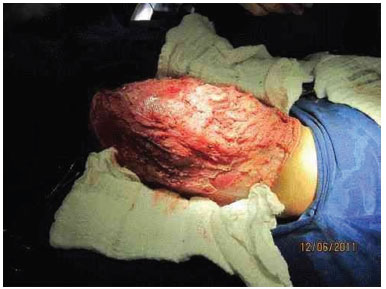
Figura 3E. Imagem do Intra operatório, durante retirada do tumor.
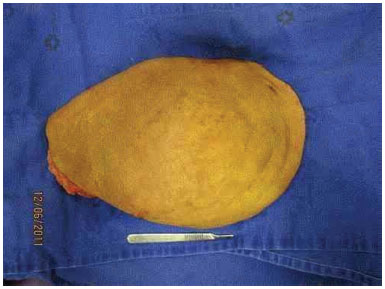
Figura 3F. Preparação para retirada do enxerto de pele para cobertura do defeito causado pela retirada da lesão.
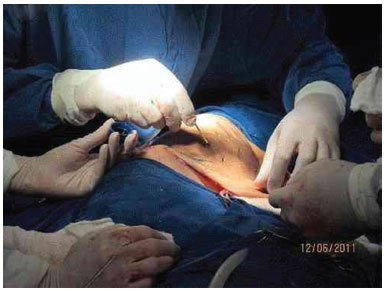
Figura 3G. Imagem da fixaçãoo do enxerto de pele.
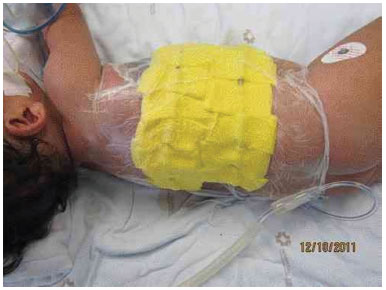
Figura 3H. Colocação do curativo à vácuo, após colocaçãoo do enxerto.
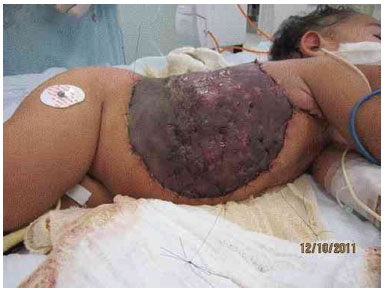
Figura 3I. Imagem do enxerto com 7 dias de pós operatório.
Com objetivo de melhorar a integração do enxerto, devido à provável drenagem de grande quantidade líquido linfático local, foi confeccionado um curativo a vácuo sobre o enxerto (Figura 3 H).
O paciente ficou sedado e entubado na UTI durante 7 dias, para manter posição adequada a fim de evitar mobilização do enxerto. Apresentou boa integração do enxerto, e evolução clínica favorável, recebendo alta hospitalar.
DISCUSSÃO
A síndrome de Proteus caracteriza-se por apresentação fenotípica polimórfica. Os achados clínicos modificam-se no decorrer do tempo, sendo a síndrome de difícil identificação. Proteus é uma síndrome rara e a história natural da doença ainda não é conhecida5. A hipótese etiológica mais aceita atualmente para a doença é genética, acreditando-se que existe mosaicismo somático e que a doença seria letal no estado não mosaico. Os casos predominantemente são esporádicos e existe relato de três adultos afetados que tiveram filhos normais9. Em alguns casos, estão presentes assimetrias e hemihipertrofias ao nascimento, mas esses achados são muito mais proeminentes na evolução pós-natal da doença. O crescimento exagerado de ossos e tecidos moles tende a um platô após a adolescência. A altura final dos pacientes, assim como seu crescimento na adolescência, parece ser normal8,11-13.
As principais sequelas da síndrome são morte prematura e ocorrência de tumores incomuns. A morte prematura deve-se principalmente à trombose venosa profunda que resulta em embolia pulmonar. Entre os tumores, os lipomas chamam a atenção por se tratarem de tumores histologicamente benignos, porém, com comportamento invasivo se intra-abdominais ou intratorácicos8.
Devido à dificuldade em diagnosticar a síndrome, critérios diagnósticos foram criados a partir da primeira conferência nacional de síndrome de Proteus em Maryland em 1998. Para fazer o diagnóstico, todos os critérios gerais devem estar presentes: distribuição em mosaico das lesões, curso progressivo e ocorrência esporádica da doença (ou seja, não familiar)8.
Inúmeros achados são possíveis na síndrome. Entre os mais comuns estão a hemi-hipertrofia, hiperostose do crânio, nevus cerebriforme, nevus pigmentado, tumores subcutâneos, malformações vasculares, tecido adiposo anormal, manifestações de sistema nervoso central e oftalmológicas diversas. Outras manifestações menos comuns são anormalidades craniofaciais, fenótipo facial, tumores, esplenomegalia, hipertrofia do timo8,9.
A hemi-hipertrofia geralmente desenvolve-se durante a infância e cessa ao final da adolescência. Pode ser parcial, completa ou cruzada, levando à inutilização do membro e dificuldade de deambulação. O tratamento é feito com epifisiodese, artrodese, encurtamento ou alongamento de membros e redução das assimetrias. Os nevus cerebriformes são lesões compostas de tecido conjuntivo com muito colágeno. Ocorrem mais comumente da superfície plantar no pé e palmar da mão. Não são critérios obrigatórios de diagnóstico, mas quando presentes são quase patognomômicos da síndrome.
Os tumores subcutâneos (lipomas, hemangiomas, linfangiomas) têm desenvolvimento variado e podem ocorrer em qualquer parte do corpo. Eles podem crescer a ponto de infiltrar tecidos locais e serem de difícil ressecção. O tratamento inclui ressecções, esvaziamentos e lipossucção. Os resultados geralmente são insatisfatórios devido à recorrência e formação de cicatrizes hipertróficas6,11. Os lipomas são compostos basicamente de adipócitos maduros. Apesar do aspecto histopatológico destes tumores ser benigno, dependendo da sua localização, principalmente se intratorácicos ou intra-abdominais, eles podem ter comportamento agressivo8. Aumento e diminuição de gorduras localizados podem ser encontrados no mesmo paciente, em diferentes partes do corpo, mostrando falta de regulação do tecido adiposo. Há um padrão que parece ser diferente dos lipomas e do aumento de gordura subcutânea. Pode ser encontrado depósito de tecido adiposo entre os músculos do pacientes14. Isto foi evidenciado em ambos nossos pacientes. No caso 1, havia infiltração de ambos os músculos grandes dorsais.
No crescimento assimétrico dos membros, principalmente os inferiores, um trabalho mostrou comparação por ressonância magnética das coxas. Frequentemente, foi encontrado aumento exagerado da gordura subcutânea do membro aumentado14. As malformações vasculares podem ter só um tipo de componente (capilar, linfática ou venosa, por exemplo) ou ser combinada (capilar e venosa ou capilar, venosa e linfática, por exemplo). Elas crescem proporcionalmente com o paciente, nunca involuem e podem expandir8. Entre os tumores menos frequentes, estão o cistoadenoma ovariano, meningioma, tumores testiculares e adenoma de parótida9.
O manejo destes pacientes deve ser feito por equipe multidisciplinar. Devem ser realizados exames a procura das principais manifestações. O acompanhamento destes pacientes deve ser prolongado devido à natureza polimórfica da síndrome. Cuidados devem ser tomados ao realizar procedimentos cirúrgicos ou imobilizações prolongadas devido à predisposição para trombose venosa profunda. Deve ser feito acompanhamento psicossocial e educacional8,9. As cirurgias vão consistir na retiradas de tumores extensos, com vascularização abundante. Foi tentado nos dois primeiros casos utilizar a lipoaspiração convencional e ultrassônica, objetivando diminuição das massas de gordura, sem lesão dos vasos. Ambos os métodos foram insuficientes, trazendo poucos benefícios estéticos e funcionais aos pacientes. Posteriormente, ambos foram submetidos à ressecção cirúrgica das massas, que se mostraram de fácil acesso, sem sangramento abundante que era o esperado, com boa evolução. É importante ter reserva de hemoderivados para o procedimento pelo risco potencial de hemorragia.
REFERÊNCIAS
1. Cohen MM Jr, Hayden PW. A newly recognized hamartomatous syndrome. Birth Defects Orig Artic Ser. 1979;15(5B):291-6. PMID: 118782
2. Wiedemann HR, Burgio GR, Aldenhoff P, Kunze J, Kaufmann HJ, Schirg E. The proteus syndrome. Partial gigantism of the hands and/or feet, nevi, hemihypertrophy, subcutaneous tumors, macrocephaly or other skull anomalies and possible accelerated growth and visceral affections. Eur J Pediatr. 1983;140(1):5-12. PMID: 6873112
3. Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16(4):899-906. PMID: 3033033 DOI: http://dx.doi.org/10.1016/S0190-9622(87)80249-9
4. Wiedemann HR, Burgio GR. Encephalocraniocutaneous lipomatosis and Proteus syndrome. Am J Med Genet. 1986;25(2):403-4. PMID: 3777031
5. Cohen MM Jr. Further diagnostic thoughts about the Elephant Man. Am J Med Genet. 1988;29(4):777-82.
6. Stricker S. Musculoskeletal manifestations of Proteus syndrome: report of two cases with literature review. J Pediatr Orthop. 1992;12(5):667-74.
7. Miura H, Uchida Y, Ihara K, Sugioka Y. Macrodactyly in Proteus syndrome. J Hand Surg Br. 1993;18(3):308-9. PMID: 8345255 DOI: http://dx.doi.org/10.1016/0266-7681(93)90047-J
8. Biesecker LG, Happle R, Mulliken JB, Weksberg R, Graham JM Jr, Viljoen DL, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84(5):389-95. PMID: 10360391 DOI: http://dx.doi.org/10.1002/(SICI)1096-8628(19990611)84:5<389::AID-AJMG1>3.0.CO;2-O
9. Cohen MM Jr. Proteus syndrome: an update. Am J Med Genet C Semin Med Genet. 2005;137C(1):38-52. PMID: 16010681 DOI: http://dx.doi.org/10.1002/ajmg.c.30063
10. Villela ALC, Guedes LGS, Paschoa VVA, David AB, Tenório TM, Lamego Junior HP, et al. Perfil epidemiológico de 58 portadores de síndrome de Klippel-Trénaunay-Weber acompanhados no Ambulatório da Santa Casa de São Paulo. J Vasc Bras. 2009;8(3):219-24.
11. Clark RD, Donnai D, Rogers J, Cooper J, Baraitser M. Proteus syndrome: an expanded phenotype. Am J Med Genet. 1987;27(1):99-117. PMID: 3605210 DOI: http://dx.doi.org/10.1002/ajmg.1320270111
12. Cohen MM Jr, Turner JT, Biesecker LG. Proteus syndrome: misdiagnosis with PTEN mutations. Am J Med Genet A. 2003;122A(4):323-4. PMID: 14518070
13. Gordon PL, Wilroy RS, Lasater OE, Cohen MM Jr. Neoplasms in Proteus syndrome. Am J Med Genet. 1995;57(1):74-8. PMID: 7645604 DOI: http://dx.doi.org/10.1002/ajmg.1320570117
14. Jamis-Dow CA, Turner J, Biesecker LG, Choyke PL. Radiologic manifestations of Proteus syndrome. Radiographics. 2004;24(4):1051-68. DOI: http://dx.doi.org/10.1148/rg.244035726
Universidade Federal do Paraná, Curitiba, PR, Brasil
Autor correspondente:
Renato da S. Freitas
Rua General Carneiro, 191
Curitiba, PR, Brasil CEP 80860-900
E-mail: dr.renato.freitas@gmail.com
Instituição: Disciplina de Cirurgia Plástica da Universidade Federal do Paraná, Curitiba, PR, Brasil.
Artigo submetido: 24/12/2011.
Artigo aceito: 24/2/2012.
 Read in Portuguese
Read in Portuguese
 Read in English
Read in English
 PDF PT
PDF PT
 Print
Print
 Send this article by email
Send this article by email
 How to Cite
How to Cite
 Mendeley
Mendeley
 Pocket
Pocket