


Case Report - Year 2019 - Volume 34 -
Neurofibromatose tipo 1 com acometimento do nervo infraorbital: relato de caso
Neurofibromatosis type 1 with infraorbital nerve involvement: a case report
Marcus Vinícius Capanema Gonçalves1,* ; Sérgio Moreira da Costa1; Liliane Carvalho Jamil1; Klaus Rodrigues de Oliveira1; Paula Pimentel Rocha Botelho1; Camila Matos Versiani1; Andreia Souto da Motta1
; Sérgio Moreira da Costa1; Liliane Carvalho Jamil1; Klaus Rodrigues de Oliveira1; Paula Pimentel Rocha Botelho1; Camila Matos Versiani1; Andreia Souto da Motta1
RESUMO
A neurofibromatose tipo 1 é uma doença autossômica dominante rara, com manifestações clínicas diversas. Sua apresentação mais marcante é a presença de neurofibromas (tumores da bainha neural) cutâneos ou internos, que também podem ocorrer de forma esporádica, associados a outras manifestações sistêmicas, como manchas café com leite e lesões oculares. Por serem tumores da bainha de mielina, os neurofibromas podem acometer diversos nervos periféricos, incluindo nervos da face. Apresentamos o caso de um paciente de 1 ano, portador de neurofibromatose tipo 1, com neurofibroma em nervo infraorbital direito, com o acesso proposto para tratamento cirúrgico que fornecesse ampla visualização e acesso a lesão, sem comprometimento estético importante, permitindo preservação de partes moles e adequado crescimento facial.
Palavras-chave: Neurofibroma; Neurofibromatose 1; Face; Pseudotumor orbitário; Órbita
ABSTRACT
Neurofibromatosis type 1 (NF1) is a rare autosomal dominant disease with multiple clinical manifestations. Its most significant presentation is cutaneous or subcutaneous neurofibromas (myelin sheath tumors), which may be associated with other systemic manifestations such as caféau- lait spots and eye involvement. Neurofibromas can affect several peripheral nerves, including the facial nerves. This report presents a case of a 1-year-old patient with NF1 with right infraorbital nerve neurofibroma in which the proposed access for surgical treatment allowed adequate visualization of the tumor with good aesthetic results, preservation of the soft tissues, and normal facial growth.
Keywords: Neurofibroma; Neurofibromatosis type 1; Face; Orbital pseudotumor; Eye socket.
INTRODUÇÃO
A neurofibromatose tipo 1 é uma doença de herança autossômica dominante, sendo uma, e a mais frequente (96%), de três doenças descritas sobre o espectro da "neurofibromatose"; as outras duas, neurofibromatose tipo 2 (3%) e schwanomatose, são clinicamente distintas do tipo 1, não sendo alvo deste trabalho1,2. É caracterizada pela presença de neurofibromas, alterações cutâneas, esqueléticas e envolvimento de múltiplos órgãos e sistemas.
Neurofibromas são tumores benignos constituídos por células de Schwann, fibroblastos, mastócitos e células perineurais, podendo ocorrer de forma esporádica como nódulos solitários, ou associados a neurofibromatose tipo 1, com nódulos solitários, múltiplos ou difusos. São a manifestação mais comum da doença, ocorrendo em até 60% dos pacientes2. Ocorrem de forma semelhante em homens e mulheres, podendo ser cutâneos ou internos, envolvendo tecidos moles profundos. Sua forma cutânea pode ser pediculada, nodular ou achatada, aumentando em número até a idade adulta2. Os neuromas internos podem acometer tecidos profundos, incluindo região periorbital, retroperitoneal, trato gastrointestinal e mediastino2. O neuroma plexiforme é a forma patognomônica da doença2, sendo constituído de neurofibromas internos que, além de crescer dentro de um nervo único, crescem envolvendo múltiplos fascículos e ramos de um nervo ou plexo.
Esse padrão de crescimento forma a característica "bag of worms" (bolsa de vermes), pela sua consistência à palpação. Diferentemente da forma cutânea, neurofibromas plexiformes tem um risco aumentado de degeneração maligna2.
Tumores malignos de nervos periféricos, neurofibrossarcomas, são de ocorrência mais rara, acometendo principalmente adultos jovens, constituindo 5-10% dos sarcomas de tecidos moles3.
Epidemiologia
A prevalência de neurofibromatose tipo 1 no mundo é de 1 caso para 3000 habitantes, podendo variar entre países e regiões, chegando até a prevalência de 1:960, em Israel1. Independente da população, 50% dos casos são decorrentes de herança familiar, e o restante de uma mutação de novo do gene causador da doença.
A expectativa de vida dos portadores da doença é reduzida em média de 8 a 21 anos, com a principal causa de morte precoce sendo o desenvolvimento de tumores malignos, que são mais frequentes nos portadores da doença, se comparados com a população geral1.
Fisiopatologia
A neurofibromatose tipo 1 é uma doença genética autossômica dominante, com uma frequência de mutações esporádicas de 50%1. É causada pela mutação no gene supressor de tumor NF1, localizado no cromossomo 17q11.22, que expressa a neurofibromina, uma proteína que regula negativamente proto-oncogenes RAS. Pela ativação de RasGTPase, a neurofibromina inativa RAS, ligando-o a sua forma inativa. A ausência dessa proteína aumenta o crescimento celular e sua sobrevivência, pela hiperativação de RAS, levando à gênese tumoral. A ausência de neurofibromina também leva a anormalidades, como lesões pigmentares, tumores cutâneos e alterações ósseas1.
Manifestações clínicas
As manifestações clínicas são variadas e podem incluir neurofibromas dérmicos e, principalmente, plexiformes, neurofibrossarcomas, anormalidades pigmentares (manifestação não neoplásica mais comum, resultantes da origem embrionária comum entre melanócitos e células de Schwann), alterações oftalmológicas, como nódulos de Lisch, glaucoma e gliomas, displasias ósseas (pela disfunção do ciclo de formação óssea por desregulação de osteoclastos e osteoblastos), além de poder envolver vários outros órgãos e provocar alterações comportamentais. Os neurofibromas, teoricamente, podem aparecer em qualquer nervo periférico. Porém, estima-se que aproximadamente 25% dos neurofibromas sintomáticos ocorram na cabeça e pescoço4.
Neurofibromas plexiformes envolvendo a pálpebra, órbita, periórbita e estruturas faciais podem levar, dentre outras alterações, a perda visual em crianças na idade de maturação visual, e se distribuem principalmente ao longo do trajeto do nervo trigêmio5. Apesar da maioria dos casos de cegueira serem secundários a gliomas no nervo óptico, neurofibromas plexiformes periorbitais também causam perda de visão, secundária a ambliopia e glaucoma5.
Diagnóstico e Tratamento
O diagnóstico da neurofibromatose tipo 1 se baseia no exame físico minucioso do paciente e na história familiar de neurofibromatose. Em casos desafiadores, como crianças abaixo de 6 anos ou sem antecedentes familiares, o estudo genético pode ser utilizado para confirmação. Visando facilitar a detecção precoce, o National Institute of Health criou uma lista de critérios diagnósticos, apresentados abaixo. Aproximadamente 30% dos pacientes portadores da doença apresentarão pelo menos um dos critérios até a idade de 1 ano; 97% dos pacientes apresentarão dois critérios até os 8 anos; e, todos os pacientes fecham os critérios diagnósticos da lista até a idade de 20 anos2,6.
Critérios diagnósticos para Neurofibromatose tipo 1
Dois ou mais dos seguintes: pelo menos seis máculas café com leite (>5mm de diâmetro em pré-púberes e > 15mm em pós-púberes); sardas em região axilar ou inguinal; glioma ótico; pelo menos dois nódulos de Lisch (hamartomas de íris); pelo menos dois neurofibromas de qualquer tipo, ou um neurofibroma plexiforme; lesão óssea distinta; parente de primeiro grau com diagnóstico de neurofibromatose tipo 12,5 (Figura 1).
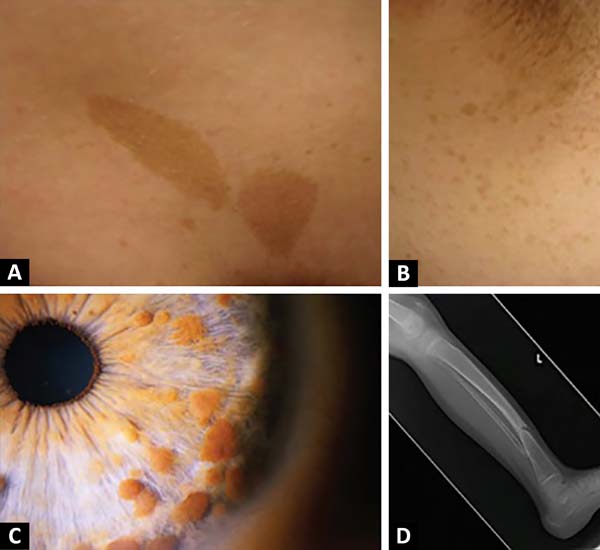
Diagnóstico diferencial deve incluir Síndrome de Legius, hiperpigmentação cutânea e tumores erroneamente diagnosticados como neurofibromas, a exemplo de alguns lipomas1.
Tem um caráter progressivo ao longo da vida, mas a severidade de progressão pode variar muito entre os acometidos.
A doença não possui um tratamento definitivo e a conduta geralmente se baseia no acompanhamento clínico e no tratamento sintomático das manifestações sistêmicas. Crianças com diagnóstico precoce devem começar acompanhamento multidisciplinar cedo e regular, com consultas oftalmológicas, dermatológicas e medidas rotineiras de pressão arterial, pela ocorrência de vasculopatia de artéria renal7,8.
Os neurofibromas dérmicos frequentemente causam prurido, dor, sangramento e problemas estéticos. Seu manejo inclui remoção cirúrgica de lesões maiores, ablação com laser de lesões menores, emolientes e suporte psicológico1,5.
Todos os pacientes portadores de NF1 que desejem ter filhos devem ser referenciados para um acompanhamento genético. Mulheres devem ser aconselhadas sobre os potenciais riscos de uma gestação, como hipertensão e a possibilidade dos neurofibromas aumentarem em número e tamanho7.
OBJETIVO
Descrever um caso de uma criança de 1 ano de idade, portadora de neurofibroma de nervo infraorbital, manifestando-se como massa expansiva e deformante em hemiface direita, e o tratamento cirúrgico com um acesso facial que promova adequada exposição da lesão, bem como possível preservação de tecidos e estética facial.
RELATO DE CASO
Paciente T.J.R.R, sexo masculino, 1 ano e 11 meses, em acompanhamento com a Dermatologia devido a presença de algumas manchas café com leite na superfície corporal e com a oftalmologia devido ao desenvolvimento de nódulos de Linch na íris (hamartomas), manifestações clínicas da neurofibromatose tipo 12. Encaminhado ao serviço de cirurgia plástica para investigação de lesão expansiva e fibrótica na região maxilar direita, com deformidade facial incluindo alargamento e levantamento da asa nasal direita, e deformidade de pálpebra inferior ipsilateral. Não apresentava qualquer acometimento visual, neurológico ou atraso no desenvolvimento neuromotor.
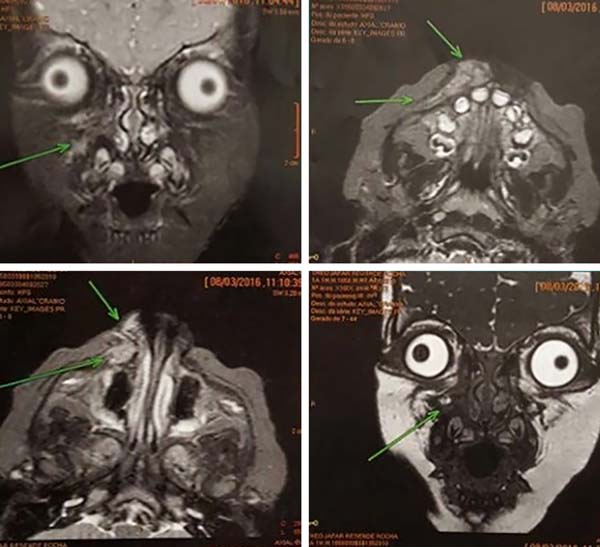
Foi solicitado uma tomografia computadorizada para melhor avaliação, que identificou massa em maxila direita, com acometimento de nervo infraorbital e alargamento do foram infraorbital, porém possivelmente sem acometimento ósseo (Figura 2). Foi então realizada biópsia incisional, que diagnosticou a massa como neuroma.

O paciente foi submetido a ressecção completa da massa por abordagem aberta, com acesso facial a Weber-Ferguson. A ressecção foi feita de modo retrógrado (do osso para a pele), na tentativa de preservar o máximo possível de partes moles (Figuras 3A e 3B). Foi ressecado o nervo infraorbital em sua origem, junto com toda a massa e tecidos adjacentes, conseguindo-se preservar um retalho de pele adequado para cobertura, assim como coxins superficiais de gordura. Não havia aparente acometimento ósseo (Figuras 4A e 4B).
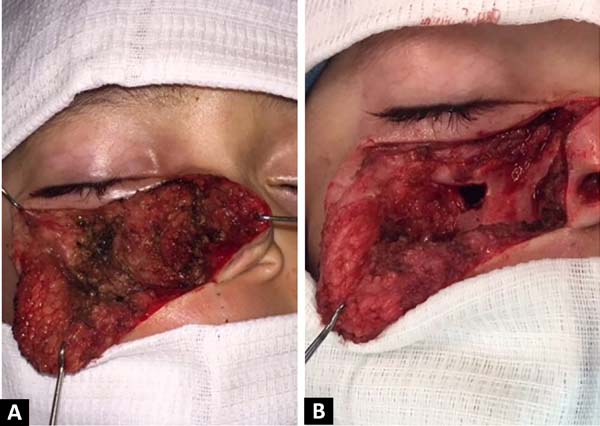
O anatomopatológico de peça cirúrgica diagnosticou a lesão como neurofibroma circundado por tecido fibrótico sem acometimento do forame infraorbital, apesar do alargamento do mesmo.
O paciente T.J.R.R. evoluiu com leve edema compatível com o pós-operatório imediato, sem complicações associadas ao procedimento cirúrgico. Está atualmente com 3 anos de pós-operatório. Apresenta possível alteração de sensibilidade da hemiface acometida no trajeto do nervo infraorbital (exame de difícil mensuração pela idade do paciente), porém sem outras sequelas ou sinais de recidiva. Apresenta bom resultado estético, com melhora da posição da asa nasal, sem retrações ou cicatrizes patológicas.
DISCUSSÃO
O tratamento cirúrgico dos neurofibromas associados a neurofibromatose tipo 1 pode ser desafiador para o cirurgião plástico, principalmente quando localizados na face, já que podem resultar não somente em deformidades de pele, mas também sequelas sensoriais e motoras pelo acometimento de nervos periféricos. Além disso, quando acometem crianças, as mesmas ainda possuem desenvolvimento e crescimento incompleto de estruturas ósseas e partes moles, tornado imperativo ressecções menos agressivas, desde que adequadas para o tratamento da patologia.
O nervo sensitivo infraorbital é a terminação do ramo maxilar do nervo trigêmeo (V par craniano), que emerge do forame infraorbital d a maxila, aproximadamente 1cm abaixo da margem orbital inferior, sendo responsável pela sensibilidade da pele da porção medial da bochecha, parte do nariz, pálpebra inferior e lábio superior9.
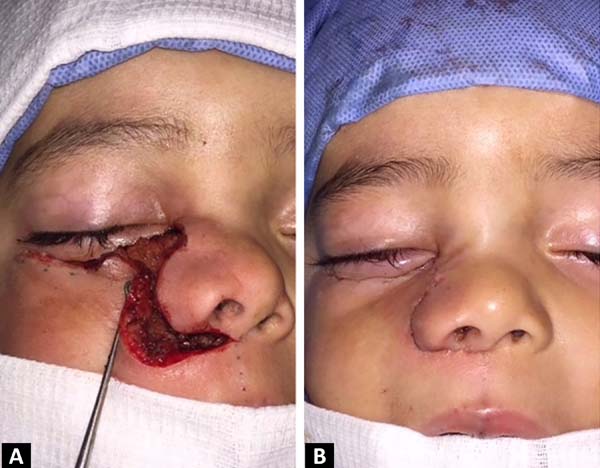
A incisão de Weber-Ferguson é um excelente acesso para tumores que envolvem maxila e órbita inferior, dando amplo acesso a essas regiões. Originalmente, o acesso pode iniciar no vermelhão do lábio superior, seguindo pelo filtro labial, ou na asa do nariz, acompanhando a parte lateral do dorso nasal e continuando para a pálpebra inferior, 3-4mm abaixo da margem ciliar. Feita a incisão em plano total, o retalho é rebatido após uma dissecção sub ou supraperiosteal, dependendo do objetivo cirúrgico e invasão tumoral10 (Figuras 5A e 5B).
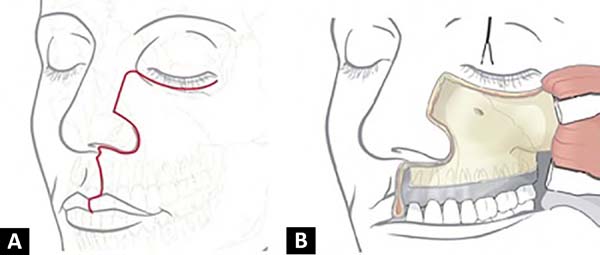

Optamos por essa incisão para adequada visualização da lesão. Iniciamos a ressecção subperiosteal de forma retrógrada até a pele, para possibilitar a preservação de partes moles não acometidas. Com ela, conseguimos ressecar toda a lesão preservando um retalho adequado de pele para cobertura, sem deformidades aparentes. Outras incisões possíveis seriam o acesso direto sobre a lesão, incisão intraoral no sulco vestíbulo-gengival ou acessos laterais.
Incisões sobre a lesão provavelmente resultariam em ressecções de pele sem necessidade, além de cicatrizes aparentes e possíveis deformidades. A incisão intraoral, por sua vez, apesar de ótima exposição óssea, não fornece adequada exposição de partes moles retrogradamente. Incisões laterais são dificultadas por dissecções amplas envolvendo parótida e ramos do nervo facial, para se ter acesso adequado à lesão.
CONCLUSÃO
Concluímos, portanto, que a abordagem cirúrgica através da incisão de Weber-Ferguson permitiu o melhor acesso para tumores de maxila envolvendo o nervo infraorbital, com ampla exposição, preservação de partes moles e resultado estético adequado, minimizando sequelas.
COLABORAÇÕES
|
MVCG |
Análise e/ou interpretação dos dados, Concepção e desenho do estudo, Investigação, Redação - Preparação do original, Redação - Revisão e Edição |
|
SMC |
Supervisão |
|
LCJ |
Investigação, Redação - Revisão e Edição |
|
KRO |
Supervisão |
|
PP |
Redação - Preparação do original |
|
CMV |
Visualização |
|
ASM |
Vizualização e revisão |
REFERÊNCIAS
1. Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017 Feb;23(3):17004. DOI: http://dx.doi.org/10.1038/nrdp.2017.4
2. Kresek JL, Walsh M. Neurofibromatosis: a review of NF1, NF2 and Schwannomatosis. J Pediatr Genet. 2016;5(2):98-104. DOI: http://dx.doi.org/10.1055/s-0036-1579766
3. Vasconcelos RAT, Coscarelli PG, Alvarenga RP, Acioly MA. Malignant peripheral nerve sheath tumor with and without neurofibromatosis type 1. Arq Neuro-Psiquiatr 2017;75(6):366-371. DOI: http://dx.doi.org/10.1590/0004-282X20170052
4. Bray DP, Chan AK, Chin CT, Jacques L. Large Cervical Vegus Nerve Tumor in a Patient with NF1 Treated with Gross Total Resection. J Brachial Plex Peripher Nerve Inj. 2016;11(1):e48-e54. DOI: http://dx.doi.org/10.1055/s-0036-1594010
5. Avery RA, Katowitz JA, Fisher MJ, Heidary G, Dombi E, Packer RJ, et al. Orbital/Periorbital plexiform neurofibromas in children with NF1. Ophthalmology. 2016 Jan;124(1):123-132. DOI: http://dx.doi.org/10.1016/j.ophtha.2016.09.020
6. Gutmann, et al. Neurofibromatosis type 1. Handbook of Clinical Neurology. 2015;132(4). DOI: http://dx.doi.org/
7. Dunning-Davies BM, Parker APJ. Annual review of children with neurofibromatosis type 1. Arch Dis Child Educ Pract. 2016;101:102-11. DOI: http://dx.doi.org/10.1136/archdischild-2014-308084
8. Hirbe AC, Gutmann DH. NF1: A multidisciplinary approach to care. Lancet Neural. 2014 Aug; 13(8):834-43. DOI: http://dx.doi.org/10.1016/S1474-4422(14)70063-8
9. Neligan P, Warren R. Plastic Surgery. 3rd ed. Philadelphia: Saunders; 2013. v. 3.
10. Colton C, Krikler S, Schatzker J, Trafton P. AO-Foundation: Weber Ferguson Midface. AO Surgery. 2019;6(4).
1. Hospital Felício Rocho, Barro Preto, Belo Horizonte, MG, Brasil.
Autor correspondente: Marcus Vinícius Capanema Gonçalves Alameda Oscar Niemeyer, 1100, 1501C, Vila da Serra, Nova Lima, MG, Brasil. CEP: 34006-065. E-mail: marcusvic1@hotmail.com
Artigo submetido: 5/1/2019.
Artigo aceito: 8/7/2019.
Conflitos de interesse: não há.
 Read in Portuguese
Read in Portuguese
 Read in English
Read in English
 PDF PT
PDF PT
 Print
Print
 Send this article by email
Send this article by email
 How to Cite
How to Cite
 Mendeley
Mendeley
 Pocket
Pocket