


Review Article - Year 2021 - Volume 36 -
Bases genômicas do carcinoma basocelular não sindrômico: revisão da literatura
Genomic bases of non-syndromic basal cell carcinoma: literature review
Daniel Sundfeld Spiga Real1,2,3,*
RESUMO
Introdução: As neoplasias cutâneas não melanoma representam o tipo mais frequente em ambos os sexos no mundo, sendo o carcinoma basocelular o mais prevalente, representando de 75 a 80% dos casos. No Brasil, o número de casos novos esperados para o triênio 2020-2022, será de 83.770 em homens e 93.160 em mulheres, correspondendo a um risco estimado de 80,12 casos novos para 100 mil homens e de 86,65 casos novos para 100 mil mulheres. Este dado demonstra a grande importância do conhecimento genômico na gênese do carcinoma basocelular esporádico.
Objetivo: Descrever os principais genes e marcadores moleculares envolvidos na predisposição e na patogênese do carcinoma basocelular não sindrômico.
Métodos: Revisão da literatura nas principais bases de dados NCBI-GTR, ClinVar, ClinGen, MedGen, OMIM e GeneReviews , utilizando como descritores: "BCC" e "basal cell carcinoma ". Critérios de inclusão: língua portuguesa ou inglesa, artigos sobre CBC esporádico. Resultados: Foram selecionados treze artigos para análise. A análise revelou uma robusta ligação da via hedgehog na gênese do carcinoma basocelular esporádico, com os principais genes envolvidos representados por PATCH1, PATCH2 e smoothened . As variantes com maior significância clínica foram SMO-M2, PTCH1 e PTCH2-?22. A mutação mais encontrada fora a relacionada à ação do UVB, sendo representada pela substituição de C>T ou CC>TT no sítio das pirimidinas, tanto no PTCH, quanto no SMO.
Conclusão: Extremamente importante aos profissionais que atuam no diagnóstico e tratamento do CBC, dentre os quais os cirurgiões plásticos, pois assim poderão melhor conduzir seus casos, com diagnósticos mais precisos e condutas de prevenção baseadas na suscetibilidade individual de cada paciente, bem como terapêuticas direcionadas e individualizadas com melhores taxas de sucesso.
Palavras-chave: Genoma humano; Carcinoma basocelular; Neoplasia de células basais; Genes; Mutação
ABSTRACT
Introduction: Non-melanoma skin neoplasms represent the most frequent type in both sexes globally, with basal cell carcinoma being the most prevalent, representing 75 to 80% of cases. In Brazil, the number of new cases expected for the triennium 2020-2022 will be 83,770 in men and 93,160 in women, corresponding to an estimated risk of 80.12 new cases for 100,000 men and 86.65 new cases for 100,000 women. This data demonstrates the great importance of genomic knowledge in the genesis of sporadic basal cell carcinoma.
Objective: To describe the main genes and molecular markers involved in the predisposition and pathogenesis of non-syndromic basal cell carcinoma.
Methods: Literature review in the main databases NCBI-GTR, ClinVar, ClinGen, MedGen, OMIM and GeneReviews, using as descriptors: "BCC" and "basal cell carcinoma". Inclusion criteria: Portuguese or EnGLIsh language, articles on sporadic BCC.
Results: Thirteen articles were selected for analysis. The analysis revealed a robust hedgehog pathway link in the genesis of sporadic basal cell carcinoma, with the main genes involved represented by PATCH1, PATCH2 and smoothened. The variants with the highest clinical significance were SMO-M2, PTCH1 and PTCH2-Δ22. The mutation most found was related to the action of UVB, being represented by the substitution of C>T or CC>TT at the pyrimidine site, both in PTCH and in SMO.
Conclusion: Extremely important to professionals working in the diagnosis and treatment of BCC, including plastic surgeons, as this way they can better conduct their cases, with more accurate diagnoses and prevention approaches based on the individual susceptibility of each patient, as well as targeted therapies and individualized with better success rates.
Keywords: Human genome; Basal cell carcinoma; Neoplasm of basal cells; Genes; Mutation.
INTRODUÇÃO
Desde tempos remotos, o maior dos enigmas representado pela origem da vida desafia filósofos e cientistas, bem como representa uma grande chave para a medicina em seus propósitos de entender, tratar e curar os males. Muitas teorias se emolduraram através dos séculos; Charles Darwin em sua emérita obra “A Origem das Espécies”1 no entrementes do ano de 1859 já dissertava sobre a importância do conhecimento sobre a constituição dos organismos para a evolução. No século seguinte, no ano de 1920, Oparin e Haldane2 apresentavam à comunidade científica a teoria dos coacervados para explicar a origem da vida e de como estruturas unicelulares derivadas de compostos orgânicos se desenvolveram para fomentar os seres e a vida como conhecemos em presentes tempos.
Conspícuo avanço no conhecimento sobre tal mote ocorreu no ano de 1990, com o início oficial do Projeto Genoma Humano, que arregimentou esforços de muitos países na busca da identificação de todos os genes humanos e suas sequências de pares de bases3. Esse projeto, que se findou no ano de 2003, proporcionou ferramentas aos pesquisadores para a compreensão de milhares de doenças, mormente as sindrômicas, bem como proporcionou uma miríade de possibilidades de ação para o tratamento e prevenção das mesmas, e o entendimento de como a substituição de única base nitrogenada poderia ocasionar consequências graves ao organismo.
Arregimentado em seus resultados, há que se ressalvarem alguns dados obtidos na primeira publicação. O genoma humano contém 3,2 bilhões de nucleotídeos, o tamanho médio dos genes é de 3.000 bases, 50% dos genes descobertos não possuem suas funções elucidadas, somente 2% do genoma codifica instruções para a síntese de proteínas, 99,9% da sequência do genoma humano é exatamente a mesma em todos os indivíduos, assim o que nos torna únicos representa 0,1% do nosso genoma. Relevante informação obtida foi a de que metade do genoma se constitui por sequências repetidas que não codificam proteínas e que não possuem funções conhecidas, mas que auxiliam na compreensão da estrutura e dinâmica dos cromossomos. Aventa-se que essas repetições possam reformular o genoma, rearranjando-o e assim criando genes novos ou modificando os já existentes3.
Tais conhecimentos emolduraram o arquétipo para a explicação de fatos observados clinicamente na prática médica, como assim se perfaz a maior incidência de carcinoma basocelular (CBC) não sindrômico em pacientes com história própria e familiar de neoplasia cutânea não melanoma. As neoplasias cutâneas não melanoma representam o tipo mais frequente em ambos os sexos no mundo, sendo o CBC o mais prevalente, representando de 75 a 80% dos casos4. No Brasil o número de casos novos esperados para o triênio 2020-2022, será de 83.770 em homens e 93.160 em mulheres, correspondendo a um risco estimado de 80,12 casos novos para 100 mil homens e de 86,65 casos novos para 100 mil mulheres. Os principais fatores de risco são: a exposição prolongada ao sol, mormente na infância e adolescência, exposição à câmara de bronzeamento artificial e história familiar de neoplasia cutânea não melanoma4.
Destarte, por representar grande relevância à saúde pública, expresso pelos números suprarretratados, faz-se necessário o entendimento das bases genômicas que propiciam essa maior pré-disposição ao desenvolvimento do CBC em indivíduos com história familiar, bem como a compreensão da ação das radiações ultravioletas nos genes, acarretando a perda de função supressora e, assim, propiciando o desenvolvimento do CBC e demais neoplasias cutâneas não melanoma. De tal forma, o presente artigo retrata o atual “estado da arte” no tangente ao conhecimento dos genes relacionados ao CBC não sindrômico, buscando individualizar medidas preventivas e terapêuticas, em conformidade com as alterações genômicas encontradas.
OBJETIVO
Descrever os principais genes e marcadores moleculares envolvidos na predisposição e na patogênese do carcinoma basocelular não sindrômico.
MÉTODOS
O desenho do estudo se perfaz como uma revisão da literatura, realizada no entrementes de outubro a novembro de 2020 nas principais bases de dados NCBI-GTR, ClinVar, ClinGen, MedGen, OMIM e GeneReviews, utilizando como descritores: “BCC” (“basal cell carcinoma”) e “basal cell carcinoma”. Figurando como critérios de inclusão: língua portuguesa ou inglesa, dados somente relacionados ao CBC não sindrômico. Já como critérios de exclusão, têm-se: línguas distintas das relacionadas nos fatores de inclusão, publicações repetidas nas diferentes bases de dados, publicações com mais de dez anos, dados sobre CBC sindrômicos - ex.: síndrome de Gorlin, síndrome do carcinoma basocelular nevoide, xeroderma pigmentoso, etc.
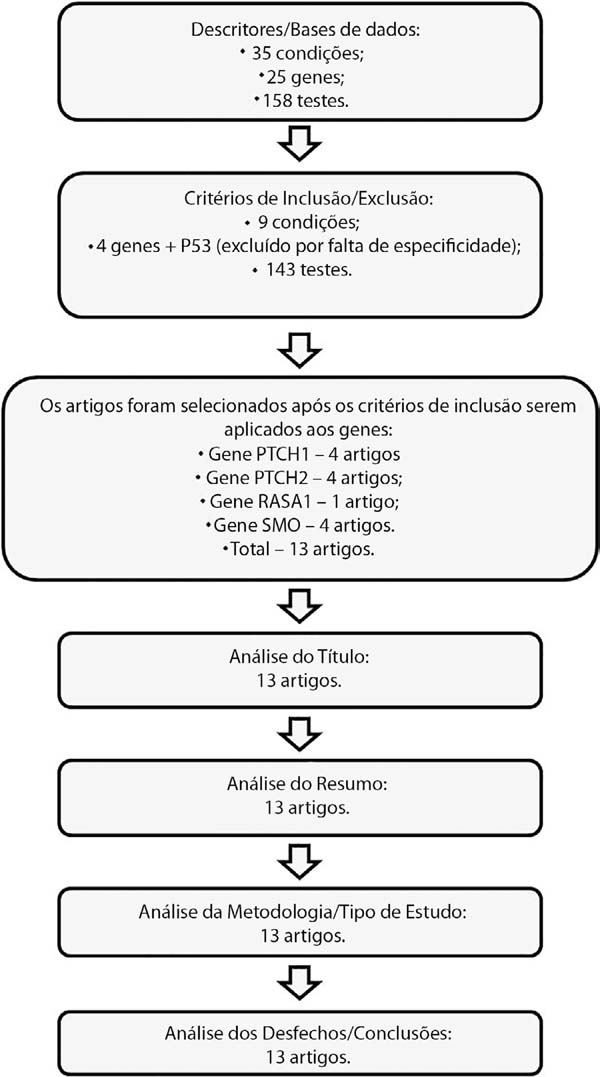
Os resultados das pesquisas foram analisados conforme fluxograma (Figura 1) para adequação aos fatores de inclusão/exclusão e posterior análise e tabulação de dados. Os artigos que contemplaram a metodologia determinada foram selecionados para o estudo e discussão. Pela heterogeneidade das bases de dados e dos resultados obtidos, os resultados foram separados conforme a base de dados em: testes, condições, genes e artigos.

RESULTADOS
Preliminarmente a busca no NCBI-GTR retornara trinta e cinco condições, vinte e cinco genes e cento e cinquenta e oito testes para os descritores utilizados. Após a primeira seleção, restaram nove condições, cinco genes e cento e quarenta e três testes.
Os artigos somente foram pesquisados após a determinação dos genes, estando incluídos os que apresentavam como mote os genes e a condição selecionados. Resultou dessa busca um total de 13 artigos que foram selecionados por cumprirem todos os critérios de inclusão e, portanto, fazendo parte de presente revisão.
Dentre as condições apresentadas, listam-se BCC1, BCC2, BCC3, BCC4, BCC5, BCC6, BCC7, BCC com diferenciação folicular e BCC sem especificações.
No tangente aos genes, retornaram o PTCH1 (patched), PTCH2 (patched), RASA1 (RAS- MAPKinase), SMO (smoothened) e o TP53 (Proteína P53). Realizando a análise de tais dados à luz dos critérios de inclusão, restou somente uma condição, mote de presente revisão - BCC1 que apresentou dados sobre os genes e os testes, pois as demais não possuem tais informações nas bases de dados até presente tempo, assim não contemplando os critérios de inclusão.
Considerando que o gene TP53 não se faz específico ao CBC, estando alterado em várias condições e síndromes, além de ter ligação somente com a condição BCC7 que foi excluída por não circunscrever os critérios de eleição, esse gene também foi excluído das análises de presente revisão. Portanto, abaixo se delineiam os quadros resumo com os artigos (Quadro 1), os testes (Quadros 2, 3 e 4) e os genes selecionados (Quadros 5, 6, 7 e 8).
| Artigos incluídos na revisão | ||||
|---|---|---|---|---|
| Referência | Ano publicação |
Tipo de estudo |
Gene | Conclusão |
| Gailani MR, et al.5 | 1966 | Experimental (amostra tumoral) | PTCH1 | O estudo demonstrou que a maior parte das mutações resultaram em proteínas “truncadas”. Espalhados por todo o gene, encontrou-se 5 prematuros “Stop Codon”, 3 mutações frameshift. Alterações que não causam proteínas truncadas - 4 mutações missense e 1 deleção. Mutações típicas de exposição solar foram encontradas por ação de UVB: substituição C-T no sítio dipirimidina, incluindo dupla base-mutação CC-TT. Uma mutação C-G transversão em um alelo fora encontrada em paciente sem história de exposição solar. O gene PTCH1 possui ação supressora tumoral, sendo uma proteína de membrana e com porção intracelular, porém sem um mecanismo ainda conhecido. |
| Aszterbaum M, et al.6 | 1998 | Experimental (amostra tumoral e sangue) | PTCH1 | O gene PTCH1 apresenta mutações somáticas no CBC. Codifica 12 domínios de proteínas transmembrana que são receptores dos ligantes da via de hedgehog. Possuem função inibitória à sinalização de tal via. Realização interação inibitória às proteínas SMO de membrana (proteínas G de membrana). No CBC há três vias para sua gênesis: mutação em proto-oncogenes - SMO e SHH (aumento de expressão) e/ou mutação com inativação do gene supressor tumoral PTCH1. CBC esporádico necessita de mutação em ambos os alelos. As mutações encontradas foram: frameshift, premature stop códon, mutações características de ação do UVB - C-T ou CC-TT foram encontradas. A maioria das mutações encontrada tiveram como consequência proteínas truncadas, principalmente no loop extracelular. |
| Zhang H, et al.7 | 2001 | Experimental (Amostra da lesão em pacientes com menos de 30 anos) | PTCH1 | As mutações encontradas foram: mudança simples de nucleotídeos, inserção de AT e deleção de 15-bp. Nucleotídeo alterados característicos de UVB C-T ou CC-TT tiveram grande prevalência no sítio da pirimidina. Na amostra somática encontradas mutações - nonsense, missense, deleção e inserção - todas com terminação prematura da proteína. Mutação do PTCH1 altera duas grandes proteínas extracelulares - loops e 12 domínios transmembranas de ligação da via hedgehog. Indivíduos jovens com mesmas alterações que indivíduos idosos, apresentam alterações em suas capacidades de reparo de DNA - p53, além de mutações no PTCH1. |
| Maglic D, et al.8 | 2018 | Experimental (amostra tumoral e sangue) | PTCH1 | A principal alteração causada pela mutações no desenvolvimento do CBC ocorre na via hedgehog, em seus reguladores - receptor PTCH1, SMO (proteína G acoplada). As mutações inibem a função repressora do PTCH1, liberando os SMO e promovendo os fatores de crescimento dos GLI intranucleares. CBC que não respondem aos inibidores de SMO, escapam de tais com mutação no sítio de ligação da droga no SMO ou com potencialização da sinalização do GLI. A sinalização Hippo também possui relação com o CBC. A via Hippo externa da hedgehog, atua através do YAP, mas não do TAZ, para iniciar a progressão do CBC, atuando na JNK-JUN (fosforilação) sinalizando; não havendo impacto na via Wnt ou hedgehog. YAP atua na cura de feridas, rápida recuperação de células basais e regeneração de epiderme. Há um aumento da YAP nuclear no CBC. YAP-TEAD-AP1 interação é requerida para iniciação e manutenção do CBC. Essa via pode ser a causa da resistência aos inibidores de SMO. |
| Smyth I, et al.9 | 1999 | Experimental (amostra tumoral e sangue) | PTCH2 | Gene supressor tumoral homólogo ao PTCH1 localizado no cromossomo 1. Presença de mutação splice no sítio doador do gene no CBC no exon 20. Não encontrada nas demais células germinativas, somente na amostra tumoral. Foi demonstrado que a inativação de um alelo do PTCH2 está relacionado ao meduloblatoma e que uma mutação em splice está presente no CBC, aventando que esteja envolvido na gênese tumoral. |
| Zaphiropoulos PG, et al.10 | 1999 | Experimental (amostra tumoral e sangue) | PTCH2 | PTCH2 é 57% idêntico ao PTCH1, com variação significante nos domínios transmembrana 6 e 7. Apresenta alternativo Splice nos exons 9 e 10 responsáveis pelo loop extracelular e os domínios transmembrana 2 e 3 da estrutura da proteína PTCH1. O PTCH2, em similaridade ao PTCH1, atua como um gene supressor tumoral. O PTCH2 possui ação distinta do PTCH1, posto que sua superexpressão não consiga compensar a inativação do PTCH1. |
| Rahnama F, et al.11 | 2004 | Experimental (amostra tumoral e sangue) | PTCH2 | PTCH2 se apresenta como um alvo da sinalização SHH-N, promovendo regulação, porém não consegui inibir ativamente o SMO-M2 contrastando com o PTCH1. PTCH2 é 57% idêntico ao PTCH1, divergindo na região hidrofílica transmembrana entre os domínios 6 e 7. O fato do PTCH1 mutado no CBC não conseguir ser compensado pela superexpressão do PTCH2, implica que o PTCH2 está relacionado a via do PTCH1, porém PTCH2 apresenta função inibitória fraca quando comparada ao PTCH1. PTCH2 apresenta múltiplas variantes, sendo as com importância clínica as PTCH2-∆22 e uma variante que Splice exons 9 e 10 e mantém os exons 8 e 11 - junção - PTCH2-∆9,10. PTCH2 em suas 3 variantes produzem proteínas no citoplasma - intracelular vesicular. PTCH2 possui a capacidade de alterar a localização dos SMO dispersos no citoplasma para um padrão de sobreposição ao PTCH1 ou PTCH2. Há a combinação do PTCH2 e PTCH1 - estão interagindo em 20% da análise de imunoprecipitado. PTCH2 atua na internalização do SHH- N. A presença da GLI1 ligada ao sítio na posição entre as bases 472-463 do códon de iniciação ATG, demonstra efeito direto do SHH sinalizador na ativação do PTCH2. A inclusão dos exons 9 e 10 e a exclusão do exon 22, são necessárias para a inibição dose- dependente. PTCH2-∆22 não diminui sua inibição ao SMO quando ligado ao SHH - demonstrando que somente o PTCH1 apresenta reposta transcricional ligante-dependente. |
| Fujii K, et al.12 | 2013 | Experimental (Amostra tumoral e Sangue) | PTCH2 | Identificado heterozigose na linhagem germinativa com mutação missense no PTCH2 na Síndrome do CBC nevoide - há um frameshift 2-bp deleção (c.1172_1173del CT no exon 9) no PTCH2 criando um prematuro stop codon - resultando em uma proteína truncada de PTCH2 - p.S391X. Não foram encontradas nenhuma mutação no PTCH1, SUFU e SMO no sangue periférico. |
| Friedman E, et al.13 | 1993 | Experimental (Amostra tumoral) | RASA1 | Encontradas três diferentes mutações no CBC no C-terminal SH2 da ras-GAP (domínio catalítico). Sugestão de que tal mutação resulte em um domínio disfuncional que não possua interação com a fosfotirosina-modificada. Há a perda da lisina na porção do SH2, perdendo a interação com fosfotirosina, existindo uma interação com grupamento amino-aromático ou causando uma hidrofobicidade. A lisina é substituída por glutamato no CBC - alterando o sítio de ligação da fosfotirosina. ras-GAP atuam nos fatores de cresimento (ex: PDGF) - família das tirosinas quinases. O sistema utilizado para detecção das mutações DGGE pode ser falho e pouco sensível. Não possui relevante ação no CBC - redução de 50% na atividade GAP não aumenta suficientemente o GTP (guanosina-trifosfato). |
| Xie J, et al.14 | 1998 | Experimental | SMO | Ativação do gene SMO através de mutação somática missense do SMO. Esse gene é um componente da sinalização do complexo SHH-receptor e traz evidências que a mutação no SMO o torna um oncogene no CBC. A ativação da via hedgehog no CBC pode resultar da falha na inibição realizada pelo PTCH ou devido a ativação anormal por uma mutação no SMO. Encontrada heterozigose de mutação missense em par de bases 1,685 - M1 alterando a Arg 562 (CGG) para Gln (CAG) - origem somática, não presente na amostra sanguínea. Há a hipótese de que o SMO mutado realize ligação com PTCH e faça que a inibição dessa cesse nos demais receptores de SMO. A sinalização do SMO mutante ocorre independente de ligantes. Há a efetiva sinalização na cascata do SHH no domínio do zinco como fator transcricional do GLI. Esse gene é amplificado nos CBC, estando com sua atividade aumentada. SMO-M2 uma proteína intrínseca da membrana, quando em superexpressão somente atua nas células basais não existindo ação em outros tecidos. SMO é um proto-oncogene, sendo um alvo para tratamento de CBC. |
| Kunstfeld R15 | 2014 | Revisão | SMO | Via hedgehog é identificada como elemento chave para o desenvolvimento de vária neoplasias. A inibição do SMO é a melhor forma de interferir na via hedgehog. A via hedgehog é fundamental no desenvolvimento embrionário e tipicamente se torna silente/inativa nos tecidos adultos. A via é iniciada com a ligação de um dos ligantes da hedgehog - Desert, Indian ou Sonic no receptor transmembrana PTCH1. Na forma inativa o PTCH exerce inibição nos receptores SMO e nenhuma sinalização interna ocorre. Quando os ligantes hedgehog se ligam na PTCH, um relaxamento na inibição ocorre e a sinalização pelo SMO se inicia, regulando a expressão e transcrição de fatores - GLI 1-3. Drogas inibidoras de SMO: Vismodegib, Sonidegib, BMS-833923(XLI139), Taladegib. Drogas que não possuem o SMO como alvo: Itraconazole (previne o cumulo de SMO no cílio primário), Vitamina D3 (ligação direta ao SMO). |
| Zhang H, et al.16 | 2018 | Revisão | SMO | Hedgehog possui importância no desenvolvimento embrionário, controle da maturação celular, diferenciação e proliferação. Nos tecidos adultos está silente, exceto na manutenção e reparo quando necessário. Os componentes da Via Hedgehog são: 3 HH ligantes (S, D e I), dois 12-transmembrana receptores - PTCH1 e PTCH2, um frizzled receptor da classe da proteína G - SMO e 3 fatores da cascata de transcrição GLI 1 -3. A transdução da sinalização HH se inicia no cílio primário - uma organela similar a uma antena que se projeta para fora das células. O efeito inibidor do PTCH inativo previne a translocação do SMO para o cílio primário. A GLI2/3 completa é negativamente regulada pela proteína SUFU e deixada na forma de GLI2/3Repressor e o sinal da via está desligado. Os ligantes HH quando se acoplam ao PTCH1, relaxam a repressão sobre o SMO e induz o cílio primário ao tráfico com SMO. Os promotores SMO ativados realizam a dissociação da GLI2/3FL do SUFU (GLI2/3R) formando o GLI2/3A - ativada, induzindo o transporte da GLI2/3A para o interior do núcleo e os gatilhos de transcrição alvo do gene. 80% dos CBC possuem mutação no PTCH1, sem alterações nos SMO, assim são responsivos aos inibidores dos SMO. Resistência ocorre com a mutação missense do SMO no lócus D473H - perda ácido aspártico. Várias mutações no sítio de ligação das drogas e nos sítios distais com redução significativa de afinidade pelo GDC-0449 (vismodegib). Há resistência causada por amplificação cromossômica da cascata dos efetores GLI2 ou uma superexpressão da via de sinalização da fosfoinoside-3-quinase. A ligação dos antagonistas de SMO induzem uma alteração conformacional tanto nos domínios transmembrana como nos domínios extracelulares, demonstrando como eles manipulam a atividade da proteína SMO. A mutação D473 causa a perda da ligação de hidrogênio, reduzindo a afinidade ao GDC-0449 ao SMO. |
| Souza AM, et al.17 | 2020 | Estudo Caso- Controle retrospectivo (Amostras pacientes com CBC e controles) | SMO | CpG-SNPs rs375350898 e rs75827493 apresentaram associação significativa ao CBC e o
SNP rs75827493 mostrara relação com o CBC nodular. Assim, tais SNPs se demonstraram
como potenciais marcadores de suscetibilidade ao CBC. A presença de SNPs na região
promotora da CpG do gene SMO, pode modificar a metilação e provocar suscetibilidade
ao CBC. CBC apresentam uma aberrante superregulação da via hedgehog, tipicamente com a perda do PTCH1 e ativação do SMO-proteína G receptor, resultando na desregulação da transcrição da GLI e processos envolvendo fatores de crescimento celular e proliferação. SNPs representam região do código que estão associadas a muitas doenças. O estudo dos SNPs nas regiões de promotores e de introns é importante para compreensão dos mecanismos da regulação genética na carcinogênese. |

| Teste para resposta a medicamentos | |
|---|---|
| Nome do teste | Laboratório |
| Basal cell carcinoma, somatic, 605462 (SMO gene) (sequence analysis-all coding exons) (postnatal) | Intergen Genetic Diagnosis and Research Centre |
| Basal cell carcinoma, somatic, 605462 (Gorlin syndrome) (PTCH2 gene) (sequence analysis- all coding exons) (postnatal) | Intergen Genetic Diagnosis and Research Centre |
| Basal cell carcinoma, somatic, 605462 (RASA1 gene) (sequence analysis-all coding exons) (postnatal) | Intergen Genetic Diagnosis and Research Centre |
| Basal cell carcinoma, somatic, 605462 (PTCH1 gene) (sequence analysis-all coding exons) (postnatal) | Intergen Genetic Diagnosis and Research Centre |
Fonte: GTR = Genetic testing registry.

| Teste para diagnóstico | |
|---|---|
| Nome do teste | Laboratório |
| PTCH1 Gene sequencing & del/dup | GeneDx |
| PTCH1 Gene sequence and deletion/duplication | Ambry Genetics |
| PTCH1 Gene sequencing and deletion/duplication analysis (PTCH1) | DDC Clinic Molecular Diagnostics Laboratory |
| Single gene testing PTCH1 | CeGaT GmbH |
| RASA1 Single Gene | Fulgent Genetics |
| PTCH1 Single Gene | Fulgent Genetics |
| PTCH2 Single Gene | Fulgent Genetics |
Fonte: GTR = Genetic testing registry.

| Teste para rastreamento | |
|---|---|
| Nome do teste | Laboratório |
| PTCH1 | Institute for Human Genetics) |
| RASA1 | Institute for Human Genetics) |
Fonte: GTR = Genetic testing registry.

| Gene PTCH 1 - Variantes exclusivas do CBC não sindrômico: cromossomo 9 (Fonte: ClinVar) |
||
|---|---|---|
| Substituição de base | Proteínas alteradas | Condições associadas |
| c.3340A>T (p.Arg1114Trp) | R1048W, R1114W, R963W, R1062W, R1113W | Basal cell carcinoma, somatic |
| 451C-T, PRO-SER | - | Basal cell carcinoma, somatic |

| Gene PTCH 2 - Variantes exclusivas do CBC não sindrômico: cromossomo 1 (Fonte: ClinVar) |
||
|---|---|---|
| Substituição de base | Proteínas alteradas | Condições associadas |
| c.3357+5C>T | - | Basal cell carcinoma, somatic |

| Gene RASA 1 - Variantes exclusivas do CBC não sindrômico: cromossomo 5 (Fonte: ClinVar) |
||
|---|---|---|
| Substituição de base | Proteínas alteradas | Condições associadas |
| c.1193G>T (p.Arg398Leu) | R398L, R221L | Basal cell carcinoma, somatic |
| c.1198A>G (p.Lys400Glu) | K223E, K400E | Basal cell carcinoma, somatic |
| c.1201A>G (p.Ile401Val) | I401V, I224V | Basal cell carcinoma, somatic |

| Gene SMO - Variantes exclusivas do CBC não sindrômico: cromossomo 7 (Fonte: ClinVar) |
||
|---|---|---|
| Substituição de base | Proteínas alteradas | Condições associadas |
| c.1604G>T (p.Trp535Leu) | W535L | Basal cell carcinoma, somatic |
| c.1685G>A (p.Arg562Gln) | R562Q | Basal cell carcinoma, somatic |

DISCUSSÃO
A heterogeneidade genética relacionada à suscetibilidade ao desenvolvimento do carcinoma basocelular denota uma grande complexidade na compreensão de todos os genes e vias de sinalizações envolvidas e representa grande desafio ao desenvolvimento de medicamentos e testes para detecção precoce e/ou pré-visualização, que sejam eficazes. Essa heterogeneidade é caracterizada por subtipos: CBC1 - ocorrendo no cromossomo 1p36, CBC2 - no cromossomo 1q42, CBC3 - 5q15, CBC4 - 12q13, CBC5 - 9p21 e o CBC6 - 7q32. Variações no início da região de transcrição da TP53 aumentam a susceptibilidade ao CBC (CBC7). Mutações somáticas, presentes somente nas lesões, não sendo encontradas nas células constitucionais dos indivíduos afetados, foram identificadas nos genes RASA1, PTCH1 e PTCH218.
Mutações são caracterizadas por qualquer alteração estável da cadeia de DNA. Podem ocorrer em três níveis diferentes: 1. molecular (genéticas ou pontuais): afetam a constituição química dos genes, ou seja, as bases nitrogenadas do DNA; 2. cromossômico: um segmento maior, envolvendo mais de um gene, é afetado, não é a constituição afetada, mas sim a estrutura; 3. genômico: mutações que afetam o conjunto do genoma, aumentando (poliploidia) ou diminuindo (haploidia ou monoploidia) o número total de jogos cromossômicos, ou alterando o número de cromossomos de cada par individual, por defeito ou por excesso19-21.
Não representando o escopo de atual revisão, será delineado breve caracterização dos principais tipos de mutações moleculares ou pontuais. Na mutação silenciosa ocorre uma mudança das bases do DNA o que leva o tripleto de nucleotídeos diferirem da sequência normal, embora codifique o mesmo aminoácido. No polimorfismo há uma alteração de uma das bases de DNA, alterando o triplito de nucleotídeos da qual faz parte, podendo ou não alterar o aminoácido correspondente. Mesmo que haja a alteração do aminoácido por um distinto do código original, há pouca ou nenhuma repercussão na função da proteína. Nos estudos arregimentados e analisados em presente revisão, as mutações relacionadas à gênese do CBC mais encontradas foram: mutações missense, nonsense e frameshift. As mutações missense se caracterizam pela alteração de uma única base do DNA, alterando o tripleto de nucleotídeo em que ela ocorre, tendo como consequência codificar um aminoácido incorreto, diferente do esperado na posição correspondente da proteína, podendo levar a alteração da função da proteína em maior ou menor grau, dependendo da localização e da importância do aminoácido. Já nas mutações nonsense também ocorre a alteração de uma única base de DNA, alterando o tripleto afetado, porém tal alteração gera um codão de terminação - “stop códon”, ou seja, a proteína nascente é truncada/cortada prematuramente, o que, dependendo do local em que ocorre, pode ou não preservar parte da função da proteína22.
Em um segundo grupo de mutações, podem-se agregar a frameshift, inserções, deleções, duplicações e expansões por repetição. Nesse grupo há uma alteração na grelha de leitura do DNA, alterando grandemente a transcrição e tradução. Nas inserções há um acréscimo de bases na sequência original do DNA, podendo ter como consequência a alteração na grelha de leitura, ou a inserção de um aminoácido “extra” o que pode alterar a função da proteína ou sua atividade. Dicotomicamente, as deleções são perdas de uma ou mais bases, perde-se um tronco de DNA, alterando a cadeia proteica que deveria ser formada e sua função. Em alguns casos, as deleções são tão extensas que podem comprometer um gene inteiro ou mesmo vários genes contíguos. Nas duplicações há um fragmento de DNA que surge copiado uma ou mais vezes relativamente à sequência original do DNA. Pode-se alterar, dessa forma, a grelha de leitura da proteína ou ocorrer a inserção de aminoácidos “extras”, que mesmo não alterando a grelha de leitura, são inadequados. Distintamente das anteriores, na frameshift, por inserção ou perda de bases, a grelha de leitura sempre é alterada. Na tradução as bases são lidas em tripletos, ou seja, cada três bases determina um aminoácido. De tal forma, ao se alterar a grelha de leitura, altera-se a forma de agrupar essas três bases e se inserem aminoácidos incorretos, com a possibilidade de se formar um “stop códon” prematuro com o truncar da proteína. Por findar, as expansões por repetição são caracterizadas por repetição de pequenas sequencias de DNA de 3 ou 4 pares de bases que se repetem em série. Mutação por expansão é uma mutação na qual o número de repetições aumentou o que pode acarretar a tradução de uma proteína com função alterada ou inativa22.
Tendo como arquétipo os conceitos supracitados, a presente revisão conseguiu delinear o atual “estado da arte” nas bases genômicas do CBC esporádico, não sindrômico. É robusta a literatura em relacionar a via de sinalização hedgehog (HH) com a gênese do CBC sindrômico e esporádico. A via HH possui extrema importância na embriogênese dos vertebrados, orquestrando o desenvolvimento, proliferação e conformação de múltiplos órgãos e sistemas. Essa via se torna silente nos tecidos adultos, somente sendo ativada quando há necessidade de reparação tecidual16. Os componentes da via HH são caracterizados por 3 HH ligantes (S, D e I), dois 12- transmembrana receptores - PTCH1 e PTCH2, um frizzled receptor da classe da proteína G - SMO e 3 fatores da cascata de transcrição GLI 1-3. A transdução da sinalização HH se inicia no cílio primário - uma organela similar a uma antena que se projeta para fora das células. O efeito inibidor do PTCH inativo previne a translocação do SMO para o cílio primário. A GLI2/3 completa é negativamente regulada pela proteína SUFU (supressor de proteína fused) e deixada na forma de GLI2/3R e, assim, o sinal da via está desligado. Os ligantes HH quando se acoplam ao PTCH1, relaxam a repressão sobre o SMO e induzem o cílio primário ao tráfico/interação com SMO - promotores. Os promotores SMO ativados realizam a dissociação da GLI2/3FL do SUFU (GLI2/3R) formando o GLI2/3A - ativada, induzindo o transporte da GLI2/3A para o interior do núcleo e os gatilhos de transcrição alvo do gene, iniciando a síntese proteica16.
A análise dos artigos selecionados para revisão (Quadro 1) revelou que os genes envolvidos na gênese, manutenção e/ou desenvolvimento do CBC são em ordem de importância: PTCH1, SMO, PTCH2 e RASA1. Outros são relacionados, mas não específicos ao CBC esporádico como PT537. O papel de tais genes no funcionamento normal da via HH está suprarretratado, já os efeitos das mutações nesses genes - criando suas variantes, abaixo será delinado. Há que se ressaltar que o gene RASA1 não possui grande comprovação na literatura, sendo ainda desconhecido o mecanismo de sua atuação na carcinogênese do CBC, estando relacionado a uma alteração no sítio de ligação de proteínas da família tirosina quinase, o que poderia intervir nos fatores de crescimento, como o PDGF, possibilitando o estímulo à proliferação celular do CBC13. Suas variantes estão relacionadas no quadro 7 e o cromossomo envolvido é o 5.
Apreende-se da literatura5-8 que o PTCH é um gene que codifica uma proteína com porções transmembrana, exterior (loop) celular e intracelular. Possui clara ação na via HH, servindo de sítio de ligação para os ligantes dessa via (indian, desert e sonic) e apresentando um papel de ativador para tal. A variante PTCH1 representa a mais importante dessas proteínas e quando está no seu estado inativo, ou seja, sem os ligantes da via HH, exerce forte inibição nos receptores SMO, impedindo que interajam com a região promotora dos cílios primários. No momento em que há ligação, no caso do CBC do ligante SHH, há uma perda na intensidade dessa inibição e a via HH é iniciada. No tangente a variante PTCH29-12, não há ainda na literatura uma completa elucidação do mecanismo de ação na carcinogênese do CBC. Apresenta-se como uma proteína homóloga ao PTCH1 com função de internalização dos ligantes SHH-N como o PTCH1. Apresenta interação com o próprio PTCH1 formando complexos, porém não possui diminuição em sua inibição ao SMO quanto acoplados aos ligantes HH. Isso determina que possuam relação com a via HH do PTCH1, porém com ação distinta. Outro aspecto que corrobora com tal proposição é o de que em mutações do PTCH1 com sua perda de função, não há uma compensação pelo PTCH2 e a gênese do CBC ocorre. PTCH2 é 57% idêntico ao PTCH1, divergindo na região hidrofílica transmembrana entre os domínios 6 e 7. PTCH2 possui a capacidade de alterar a localização dos SMO dispersos no citoplasma para um padrão de sobreposição ao PTCH1 ou PTCH211. Os cromossomos envolvidos são o cromossomo 9 no PTCH1 e o cromossomo 1 no PTCH2.
Em referência as variantes desses genes, a alteração mais encontrada foi relacionada à ação do UVB nas células somáticas das lesões, ou seja, uma substituição (missense) de C>T ou CC>TT nos sítios das pirimidinas. Outras mutações, com menor prevalência e menores significados clínicos, também foram descritas e estão resumidas nos quadros 5 e 6, respectivamente dos genes PTCH1 e PTCH2. Há que se ressalvar que o PTCH2 apresenta múltiplas variantes, porém as que possuem significância clínica são três: PTCH2; PTCH2-∆22 (exclusão do éxon 22) e PTCH2-∆9,10 (inclusão dos éxons 9 e 10 por splice). Por tal, as variantes determinam a gênese do CBC esporádico por perderem suas funções de supressoras da via HH, acarretando na ativação da via e sua cascata nuclear e conseguinte proliferação celular basal.
O gene com maior importância para a farmacogenética se representa pelo SMO. Está localizado no cromossomo 7 e suas variantes relacionadas no Quadro 8. Configura-se como o principal alvo para medicamentos que visam o tratamento dos CBCs que não respondem às terapias tradicionais23-27. São responsáveis pelo transporte dos ligantes HH para o interior celular através de sua interação com os cílios primários, representam os verdadeiros ativadores da via HH, com atividade constante sem o efeito inibidor do PTCH. Por tal, são considerados proto-oncogenes, ou seja, quando sofrem mutação e não mais são inibidos pela PTCH, ativam a via HH e se autoalimentam com a inibição do PTCH, realizando a gênese tumoral, assim caracterizados como oncogenes14-17. A sinalização do SMO mutante ocorre independente de ligantes. Há a efetiva sinalização na cascata do SHH no domínio do zinco como fator transcricional do GLI. Esse gene é amplificado nos CBC, estando com sua atividade aumentada. SMO-M2, uma variante com grande significância clínica, proteína intrínseca da membrana, quando em superexpressão somente atua nas células basais não existindo ação em outros tecidos. SMO é um proto-oncogene, sendo um alvo para tratamento de CBC14.
Os promotores SMO ativados realizam a dissociação da GLI2/3FL do SUFU (GLI2/3R) formando o GLI2/3A - ativada, induzindo o transporte da GLI2/3A para o interior do núcleo e os gatilhos de transcrição alvo do gene. A GLI2/3 completa é negativamente regulada pela proteína SUFU e deixada na forma de GLI2/3R (inativa/repressor) e o sinal da via fica em estado silente/desligado16. As variantes do SMO são SMO-M1 e SMO-M2 (esta sendo a que possui importância clínica e terapêutica). Tais variantes apresentam a capacidade de ativas da via HH sem que haja ligantes HH nos promotores dos receptores, assim ocorre uma amplificação da via, isso associada a uma perda da inibição do PTCH, como acima descrito, resultando no desenvolvimento do CBC esporádico. O SMO é um receptor “proteína G-acoplado” (família frizzeled)28, estando relacionados como possíveis marcadores de suscetibilidade ao desenvolvimento de CBC, além de serem os melhores alvos para as terapias farmacológicas. CpG-SNPs rs375350898 e rs75827493 apresentaram associação significativa ao CBC e o SNP rs75827493 mostraram relação com o CBC nodular. Assim, tais SNPs se demonstraram como potenciais marcadores de suscetibilidade ao CBC. A presença de SNPs na região promotora da CpG do gene SMO, pode modificar a metilação e provocar suscetibilidade ao CBC. Esses apresentam uma aberrante superregulação da via hedgehog, tipicamente com a perda do PTCH1 e ativação do SMO-proteína G receptor, resultando na desregulação da transcrição da GLI e processos envolvendo fatores de crescimento celular e proliferação17.
A miríade de interações necessárias para a gênese neoplásica pode ser retratada por novas vias relacionadas à via HH nas bases genômicas do CBC esporádico. Um exemplo é encontrado na via Hippo (YAP-TED-AP1) que possui atuação diretamente nos promotores GLI intranucleares e parece estar relacionada à resistência de alguns CBC aos inibidores de SMO8, mas ainda não há estudos suficientes para confirmação desses achados até presente momento.
Limitações do estudo
Emoldurado como uma revisão da literatura não sistemática, o presente apresenta os vieses e fragilidades inerentes desse tipo de estudo. Outrossim, os estudos analisados são, em sua maioria, experimentais sem grupo controle e com análise de pequeno número de amostras, bem como não há uma homogeneidade e randomização dos participantes, ou seja, não se realizaram distinção de etnia, Fitzpatrick, gênero, idade, exposição ambiental, origem, laboro, utilização de medicamentos (ex.: hidroclorotiazida), comorbidades, etc. Tais considerações podem acarretar vieses e prejudicar a análise. Os próprios métodos de análises da mutação utilizados na maioria dos estudos não possuem boa sensibilidade para detecção de alguns tipos de variações, prejudicando a análise de presente revisão.
CONCLUSÃO
O CBC esporádico representa iminente problema de saúde, representado o tipo mais prevalente de neoplasia e causando grande impacto no sistema público de saúde. A presente revisão apresenta robusta literatura, demonstrando a importância do conhecimento genômico dessa moléstia para melhores terapêuticas, prevenção e rastreamento de suscetibilidade. Medicamentos desenvolvidos baseados nesses conhecimentos se apresentam como importantes meios de tratamento aos pacientes que não possuem condições cirúrgicas e/ou não respondem à radioterapia. Nesse aspecto, medicamentos como o vismodegib, o itraconazol, a vitamina D3, bem como outros em fase I e II de estudo23-27 serão responsáveis pela cura desses pacientes em futuro breve, além de medicamentos imunomoduladores e de terapia gênica.
Por todo o suprarretratado, é extremamente importante aos profissionais que atuam no diagnóstico e tratamento do CBC, entre os quais os cirurgiões plásticos, essenciais e os mais desafiados por essa condição, arregimentarem os conhecimentos sobre as bases genômicas dessa patologia, pois assim poderão melhor conduzir seus casos, com diagnósticos mais precisos, e condutas de prevenção baseadas na suscetibilidade individual de cada paciente, bem como terapêuticas direcionadas e individualizadas com melhores taxas de sucesso.
COLABORAÇÕES
|
DSSR |
Análise e/ou interpretação dos dados, Análise estatística, Aprovação final do manuscrito, Coleta de Dados, Conceitualização, Concepção e desenho do estudo, Gerenciamento de Recursos, Gerenciamento do Projeto, Investigação, Metodologia, Realização das operações e/ ou experimentos, Redação - Preparação do original, Redação - Revisão e Edição, Supervisão, Visualização |
REFERÊNCIAS
1. Browne J. A origem das espécies de Darwin: uma biografia. Rio de Janeiro: Editora Zahar; 2007.
2. Dick SJ. The biological universe. Netherlands: Springer; 1999.
3. Department of Energy, Genomics and Its Impact on Science and Society (US). Human genome program project information archive - 1990-2003 [Internet]. Washingtion: 2008. Disponível em: http://www.ornl.gov/sci/techresources/Human_Genome/publicat/primer2001/4.shtml
4. Instituto Nacional de Câncer José Alencar Gomes da Silva (INCA). Estimativa 2020: incidência de câncer no Brasil. Rio de Janeiro: INCA; 2019.
5. Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, Pressman C, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996 Set;14(1):78-81.
6. Aszterbaum M, Rothman A, Johnson RL, Fisher M, Xie J, Bonifas JM, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol. 1998 Jun;110(6):885-8.
7. Zhang H, Ping XL, Lee PK, Wu XL, Yao YJ, Zhang MJ, et al. Role of PTCH and p53 genes in early-onset basal cell carcinoma. Am J Pathol. 2001 Fev;158(2):381-5.
8. Maglic D, Schlegelmilch K, Dost AF, Panero R, Dill MT, Calogero RA, et al. YAP-TEAD signaling promotes basal cell carcinoma development via a c-JUN/AP1 axis. EMBO J. 2018 Set;37(17):e98642.
9. Smyth I, Narang MA, Evans T, Heimann C, Nakamura Y, Chenevix-Trench G, et al. Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene in basal cell carcinoma and medulloblastoma on chromosome 1p32. Hum Mol Genet. 1999 Fev;8(2):291-7.
10. Zaphiropoulos PG, Undén AB, Rahnama F, Hollingsworth RE, Toftgård R. PTCH2, a novel human patched gene, undergoing alternative splicing and up-regulated in basal cell carcinomas. Cancer Res. 1999 Fev;59(4):787-92.
11. Rahnama F, Toftgård R, Zaphiropoulos PG. Distinct roles of PTCH2 splice variants in Hedgehog signalling. Biochem J. 2004 Mar;378(Pt 2):325-34.
12. Fujii K, Ohashi H, Suzuki M, Hatsuse H, Shiohama T, Uchikawa H, et al. Frameshift mutation in the PTCH2 gene can cause nevoid basal cell carcinoma syndrome. Fam Cancer. 2013 Dez;12(4):611-4.
13. Friedman E, Gejman PV, Martin GA, McCormick F. Nonsense mutations in the C-terminal SH2 region of the GTPase activating protein (GAP) gene in human tumours. Nat Genet. 1993 Nov;5(3):242-7.
14. Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, et al. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998 Jan;391(6662):90-2.
15. Kunstfeld R. Smoothened inhibitors in the treatment of advanced basal cell carcinomas. Curr Opin Oncol. 2014 Mar;26(2):184-95.
16. Zhang H, Sun Z, Liu Z, Song C. Overcoming the emerging drug resistance of smoothened: an overview of small-molecule SMO antagonists with antiresistance activity. Future Med Chem. 2018 Dez;10(24):2855-2875.
17. Souza AM, Lopes OS, Liberato AL, Oliveira PJR, Herrero SST, Nascimento ALD, et al. Association between SNPs and loss of methylation site on the CpG island of the promoter region of the smoothened gene, potential molecular markers for susceptibility to the development of basal cell carcinoma in the Brazilian population. Asian Pac J Cancer Prev. 2020 Jan;21(1):25-29.
18. Hamosh A, Ramussen SA; Online Mendelian Inheritance in Man (OMIM). Basal cell carcinoma, susceptibility to 1 BCC1 [Internet]. Baltimore: OMIM/Johns Hopkins University; 2000. Disponível em: https://omim.org/entry/605462?search=BCC&highlight=bcc
19. Leroi A. Mutants: on the form, varieties & errors of the human body. London: HarperCollins; 2003.
20. Maki H. Origins of spontaneous mutations: specificity and directionality of base-substitution, frameshift, and sequence-substitution mutageneses. Annu Ver Genet. 2002;36:279-303.
21. Taggart R. Starr C. Biology the unity and diversity of life: mutated genes and their protein products. Pacific Grove: Thompson Brooks/Cole; 2006.
22. Universitat de Barcelona (ES). Sant Joan de Déu - Hospital Materno Infantil. Guía metabólica - tipos de mutações [Internet]. Barcelona: Sant Joan de Déu. Disponível em: https://metabolicas.sjdhospitalbarcelona.org/sites/default/files/tipos_de_mutacoes_ptg.pdf
23. Lacroix C, Fish I, Torosyan H, Parathaman P, Irwin JJ, Shoichet BK, et al. Identification of novel smoothened ligands using structure-based docking. PLoS One. 2016 Ago;11(8):e0160365.
24. Byrne EFX, Sircar R, Miller PS, Hedger G, Luchetti G, Nachtergaele S, et al. Structural basis of smoothened regulation by its extracellular domains. Nature. 2016 Jul;535(7613):517- 522.
25. Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015 Mar;27(3):342-53.
26. Pricl S, Cortelazzi B, Dal Col V, Marson D, Laurini E, Fermeglia M, et al. Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma. Mol Oncol. 2015 Fev;9(2):389-97.
27. Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell. 2015 Mar;27(3):327-41.
28. GeneCards - The Humam Gene Database. Homepage [Internet]. Rehovot: Weizmann Institute of Science; 1996. Disponível em: https://www.genecards.org/cgi-bin/carddisp.pl?gene=SMO
1. Universidade Federal de São Carlos, Programa de Pós-Graduação - PPGBiotec, São
Carlos, SP, Brasil.
2. Sociedade Brasileira de Cirurgia Plástica, São Paulo, SP, Brasil.
3. Instituto Sundfeld de Cirurgia Plástica, São Carlos, SP, Brasil.
Autor correspondente: Daniel Sundfeld Spiga Real, Rua Dr. Domingos Faro, nº 285 - Jardim Alvorada, São Carlos, SP, Brasil, CEP 13562-003. E-mail: dplasticsurgery@hotmail.com
Artigo submetido: 26/11/2020.
Artigo aceito: 23/04/2021.
Conflitos de interesse: não há.
 Read in Portuguese
Read in Portuguese
 Read in English
Read in English
 PDF PT
PDF PT
 Print
Print
 Send this article by email
Send this article by email
 How to Cite
How to Cite
 Mendeley
Mendeley
 Pocket
Pocket